
記住我


Signal transduction is heavily reliant on the activity of protein kinases which, by catalyzing protein phosphorylation, control many cellular processes, regulating intercellular communication and coordination of complex functions.
In humans, the complex regulation of processes like immunity, neurobiology, cell cycle or morphogenesis is ensure by 518 protein kinases and approximately 20 lipid kinases that compose the human kinome (∼1.7% of human genes).1,2 478 of the 518 human protein kinases contain a eukaryotic protein kinase (ePK) domain, shared by all the classical kinase families.3,4 The ePKs are further classified into eight major groups (AGC, CAMK, CK1, CMGC, STE, TK, TKL and RGC kinase families) based on sequence similarity within this domain and can be roughly divided in those able to phosphorylate tyrosine residues and those able to phosphorylate the serine or threonine residues (also known as STK). Approximately 350 of ePKs belong to the STK group and are primarily involved in transmitting the signals within the cell, amplifying and modulating the signals received from the local microenvironment.5 One of the principal groups of STK, highly conserved across organisms, is the CMGC kinase family, named after the initials of its subfamily members, including cyclin-dependent kinase (CDK), mitogen-activated protein kinase (MAPK), glycogen synthase kinase (GSK) and CDC-like kinase (CLK). In total CMGC kinase family has 63 family members, including also CMGC subfamilies DYRK and SRPK.3,6,7
Here we focus on the roles of CDKs in physiological and pathological contexts. CDKs were first identified as the kinases able to regulate the progression through the different phases of the cell cycle in association with their activating partners cyclins. However, we now know that the 20 CDKs present in the human genome can be divided in at least three different sub-family and that, during the evolution, each CDK has acquired specific and distinct functions that extend far beyond the “simple” regulation of cell cycle progression.8
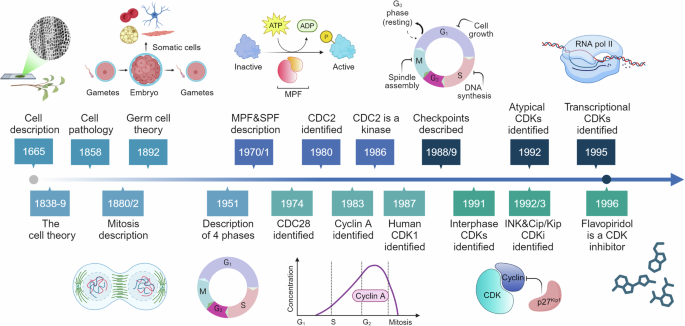
The long journey from the first description of the cell to the clinical use of CDKs inhibitorsThe development of the first optical microscopes at the end of XVI century provided a significant boost to the study of biological and medical sciences. In 1665, physicist Robert Hooke observed, using one of the first microscopes, small, distinct structures in elderberry pith, which he named “cells” due to their resemblance to the rooms occupied by monks in convents. Hooke was actually observing dead cells and therefore only described their walls, not the nucleus or organelles9 (Fig. 1).
Fig. 1
From the cell discovery to the CDKs targeting: a subjective timeline. Timeline of milestone events leading to the discovery, research and development related to cyclin dependent kinases (CDKs), cyclins and cell cycles regulators. In this long journey many more milestones could be identified, and this timeline represents our personal point of view. See text for references; caption of the original cork picture, described in “Micrographia” by Robert Hooke (1665), is reported in the Figure. (Adapted from “Timeline (7 segments, Horizontal), by BioRender.com (2024). Retrieved from https://app.biorender.com/biorender-templates)
It took about 200 years of crucial observations during the XVII and XVIII centuries to confirm the presence of the cells in living organism. This culminated in the development of the “cell theory” by Matthias Schleiden and Theodor Schwann in 1838-1839, which asserted that all plants and animals are composed of cells.10,11 Then in 1858 Rudolf Virchow proposed that every cell arises from another pre-existing cell (“Omnis cellula e cellula”) and established the foundation for cell pathology as a scientific discipline.12
In 1880/2 advancements in staining techniques and microscopy allowed Walther Flemming and Eduard Strasburger to describe the process of mitosis, identify chromosomes, and distinguish the phases of cell division.13,14 In the next decade August Weisman suggested that chromosomes were the basis of heredity, proposing that genetic information is stored and transmitted by chromosomes in germ cells. He also proposed that somatic cells had limitation in their reproductive potential, which might explain ageing and natural death.15,16
These illuminating works on the mitotic division and chromosomes segregation were confirmed in the first 20 years of XX century but then “the period between 1920 and 1950 was somewhat of a Dark Ages for the cell cycle”.17 However, the work in these years laid the foundations for the description of the Mitosis four phases one of synthesis (S phase) and one of division (M phase) interspersed by two phases of Gap (G1 and G2) described by Howard in 1951.18,19
After the cell cycle was established as consisting of G1, S, G2, and M phases subsequent studies focused on understanding how transitions between these phases are regulated and led to the discovery of the M phase- and an S phase-promoting factor (MPF and SPF) which accelerate the onset of mitosis and S phase progression, respectively.17,19
The introduction of genetics to cell cycle studies by researchers like Hartwell and Nurse, led to the identification in yeast cells of cell division cycle (CDC) genes as necessary for cell cycle progression. Temperature sensitive lethal mutants allowed the identification of CDC28 in budding yeast as a gene necessary to control the first step of cell cycle progression in G1 phase and of CDC2 proteins in fission yeast as necessary for G2/M transition.17,20,21 Subsequently, using complementation approaches, the eucaryotic homologs of CDC2 (now named CDK1) has been identified and has been introduced the concept that CDKs are kinases regulated by post translational modifications.17,22,23,24 In the same years by studying the cell division of sea urchin eggs Hunt and collaborators identified one protein destroyed every time the cells divide that they proposed to name cyclin (i.e. cyclin A).25 At the end of 80 s, the concept that specific checkpoints exist to ensure a correct progression along the cell cycle and that damaged DNA is not transmitted to daughter cells was developed.26 The 90 s could marked a deeper understanding of the complex regulation of cell cycle progression, beginning with the identification of interphases cyclins and CDKs and with the key roles of TP53 and Retinoblastoma proteins as major targets and regulators of CDKs activity and cell cycle progression.27,28,29,30 A significant advancement was the identification of a new class of cell cycle regulating proteins: the CDK inhibitors (CKIs). CKIs inhibit the activity of CDKs by either preventing CDK4 and CDK6 binding to cyclins (INK family) or by inhibiting the activity of formed cyclin-CDK complexes (Cip/Kip family), recently reviewed by others.31
In the same years Harlow and collaborators, using degenerate oligonucleotides corresponding to conserved regions of CDC2 (CDK1) to amplify human sequences via PCR, identified five other genes clearly related to CDK1.32 Similarly, others researchers looking for the expression of CDK1-like genes in human neurons identified CDK5.33,34 These observations introduce the concept that CDKs form a large family of serine-threonine kinases that could have diverse functions and could be expressed in a tissue-specific manner. The CDKs family continued to expand with the identification of CDK7 (also known as a CDK Activating Kinase or CAK),35,36 CDK837 and CDK9,38 now categorized as transcriptional CDKs (see next paragraphs).
CDKs play a role in various pathological conditions (as discussed in the following paragraphs). It is no surprise that their discovery stimulated the research of compounds able to inhibit their activity. This successful line of research, started with the identification that flavopiridol, a flavone with antitumor activity, that was the first in class molecule able to inhibit all cell-cycle related cyclin-CDK complexes.39 Although flavopiridol lacks the necessary specificity and potency to become a drug with a possible clinical use, we can say that its identification as CDK inhibitor open a new era in the treatment of several type of human cancers, as better described below (see section “CDKs as therapeutic targets”). The journey to understanding how a single cell divides into two daughter cells with high fidelity in transmitting genetic information was long and marked by the accumulation of experimental evidence, the formulation of new ideas and theories, and significant technical advancements (Fig. 1). This progress has ultimately paved the way for a new era of precision medicine, particularly in cancer treatment, where specific CDKs can now be precisely targeted for more effective patient care.
CDK subfamilies expression and regulation in normal tissueIn the human genome are present 21 genes encoding 20 CDKs, named from CDK1 to CDK20, with CDK11 encompassing two isoforms, encoded by separate genes (CDK11A and CDK11B). CDKs can be classified into three phylogenetic subgroups based on their primary functions: 1) primarily involved in cell cycle regulation (CDK 1, 2, 3, 4, and 6); 2) primarily involved in transcription regulation (CDK 7, 8, 9, 10, 11, 12 and 13); and 3) atypical CDKs (CDK 5, and 14–20).8 Each CDK has acquired specific and distinct functions beyond merely regulating cell cycle progression (Fig. 2).
Fig. 2
Generation and Analysis of the Phylogenetic Tree of Human Full-Length CDKs. The phylogenetic tree of human full-length CDKs was generated using the ggtree R package. Homology among the protein sequences was evaluated using Multiple Sequence Alignment (MSA) analysis, with CDK12 and CDK13, the longest CDKs, serving as references. The MSA reference number denotes the alignment position. Different amino acids in the protein sequences are color-coded, and gaps are introduced to properly separate conserved amino acid regions among CDKs (indicated by horizontal lines/spaces). The Serine/Threonine protein kinase domain is well conserved across all CDKs, although they show significant divergence in the lengths of their N-terminal and C-terminal extensions. On the right, a schematic representation of CDKs is provided, highlighting the kinase domain and indicating the amino acid length of each CDK. (Created with BioRender.com)
While cell cycle CDKs, with the exception of CDK3, are well studied and characterized, much less is known about the regulation and biological function of transcriptional and atypical CDKs (with the exception of CDK5). These CDKs remain part of the Dark Kinases group, which include nearly one-third of identified human kinases, and are listed in the Dark Kinase database (DKK; https://darkkinome.org). In the following sections, we will provide a detailed description of how the three CDKs subfamily are regulated by their most relevant binding partners and phosphorylation events (Table 1).
Table 1 Overview of CDKs-cyclin complexes and their main regulatorsAmong understudied kinases, there is also the related family of CDKL (CDK like Kinases), composed of five members CDKL1 to CDKL5 which have high sequence similarity to CDKs and encode for a cyclin binding domain, although there are no evidence of their interaction with cyclins (reviewed in Ref. 40). The members of CDKL family will not be discussed here.
Cell cycle CDKsThe eukaryotic cell cycle (Fig. 3) can be defined the universal process by which cells correctly divide into two daughter cells ensuring that no damaged DNA is transmitted. This process underlies the growth and development of all living organs.17
Fig. 3
Cell Cycle Progression via EGFR and Receptor Tyrosine Kinase Signaling. EGFR stimulation promotes the activation of the CycC-CDK3 complex, enabling the cell to exit quiescence (G0) and enter the G1 phase by priming RB phosphorylation. Activation of various Receptor Tyrosine Kinases (RTKs) can similarly promote G1 progression through signal cascades involving RAS-GTP and its downstream RAF/MEK/ERK and PI3K/AKT/mTOR pathways. These signaling cascades lead to the formation and activation of CycD-CDK4/6 complexes and their translocation to the nucleus. In the nucleus, CycD-CDK4/6 complexes further phosphorylate RB. The inhibition of RB allows the accumulation of E2F on DNA, promoting the transcription of genes essential for cell cycle progression and DNA replication. Subsequently, the activation of the CycE-CDK2 complex drives the transition from G1 to S phase by hyper-phosphorylating RB, enabling cell cycle progression independently of growth factor stimuli (bypassing the restriction point). The accumulation of CycA and the displacement of CycE from the CycE-CDK2 complex facilitate the formation of the CycA-CDK2 complex, which drives S phase entry, progression, and DNA synthesis. Following faithful DNA replication, the CycA-CDK1 complex triggers entry into mitosis. This is followed by the formation and activation of the CycB-CDK1 complex, which is necessary for the completion of proper cell division. The roles of activating proteins (such as CAK and CDC25A/B/C) and inhibitory proteins (such as CDK inhibitors, WEE1, and MYT1) on specific cyclin-CDK complexes are indicated by black arrows. Abbreviations used: RTKs Receptor Tyrosine Kinase, CAK complex CDK Activating Kinase complex, CycA cyclin A, CycB cyclin B, CycC cyclin C, CycD cyclin D, CycE cyclin E, CDC25 Cell Division Cycle 25. (Adapted from “Cell Cycle Checkpoints”, “RAS Pathway”, by BioRender.com (2024)
Cell cycle goal is to guarantee that DNA faithfully replicates once during S phase (DNA Synthesis phase) and that identical chromosomes copies segregate equally into two dividing cells during mitosis (or M phase).17 Progression along the different phases of the cell cycle is strictly monitored to ensure that S phase is completed before mitosis begins. There are two Gap phases in somatic cell cycle: G1 separating the M and S phase, and G2 separating the S and M phase- absent in early embryonic cell cycles. During G1 cells respond to extracellular stimuli that make the cell decide whether to replicate its DNA and divide or to exit the cell cycle and enter a quiescent state (G0). Once entered into the mitotic cell cycle and decide to replicate their DNA, cells are irreversibly committed to complete the cycle and this decision is virtually taken by overstepping the so called “restriction point” or G1 checkpoint, set in late G1.41 While continuous mitogenic stimulation is necessary for cells to cross the restriction point, once this checkpoint is left behind, the cell cycle takes place in a mitogen independent manner. Similarly anti-proliferative signals can exert their effect only if they target the cell in early G1 before passing through the restriction point. Yet, if damaged or incompletely replicated DNA is not sufficiently repaired cells block the cell cycle in G2 and do not initiate mitosis thanks to the activation of the G2/M checkpoint, linking DNA damage response to cell cycle progression.42,43
To ensure that cell cycle progress and arrest as necessary, cell cycle CDKs, including CDK1, CDK2, CDK3, CDK4 and CDK6 (reviewed in Ref. 8), are usually constitutively expressed in quiescent cells as inactive form and, their activity tightly regulated by a four-level activating cascade that include environmental factors, cyclins expression, CDKs phosphorylations and CDKs inactivation (Fig. 3). Schematically:
I.Cell decision to progress through the cell cycle and divide is based on its ability to sense and transduce environmental signals like nutrients, mitogens, or cytostatic factors. For instance, growth factor stimulation (e.g., EGF) activates the RAS-RAF-MEK-ERK1/2 pathway, a crucial signal transduction cascade that drives cells from a quiescent state (G0) into the cell cycle, mainly by regulating cyclins and CKI expression. Once cells commit to entering the cycle, they are irreversibly committed to completing it, regardless of the continued presence of these external signals.44
II.Each cell cycle phase is characterized by the expression of specific cyclins, which regulate CDK activation. Cyclin levels fluctuate throughout the cycle, with their synthesis and degradation (known as “cyclin waves”) tightly controlling kinase activity in a time-dependent manner.8 Cyclins not only activate CDKs but may also influence CDK substrate specificity. Ten cyclins, grouped into four types (A-, B-, D-, and E-type), associate with cell cycle CDKs.8,45
III.The final step in fully activating cyclin-CDK complexes involves phosphorylation by the CAK complex (CDK Activating Complex) in the nucleus. The CAK complex is a heterodimer formed by the catalytic subunit CDK7, the regulatory subunit cyclin H and the assembly factor MAT1. Several evidences suggest that cyclin binding is necessary for CAK phosphorylation, as it induces conformational changes in the CDK, exposing the phosphorylation site within the T-loop. This allows way, the CAK complex to phosphorylate and activate CDKs (CDK1 at T161, CDK2 at T160, CDK3 at T161, CDK4 at T172, and CDK6 at T177).35,46
IV.CDK inactivation occurs through phosphorylation on specific sites or by binding with CDK inhibitors (CKIs). Phosphorylation of threonine and tyrosine residues in the G-loop (T14 and Y15), by WEE1 and MYT1 inhibit kinase activity, preventing cell cycle progression, particularly in response to DNA damage. This phosphorylation does not significantly alter CDK structure but reduces its substrates affinity.8,47,48
In the second case two families of CKIs have been defined. INK4 proteins (inhibitors of CDK4), including p16INK4a, p15INK4b, p18INK4c and p19INK4d, which specifically inhibit CDK4 and CDK6 and the CIP/KIP family (i.e. p21Cip1/Waf1/Sdi1, p27Kip1, and p57Kip2), which bind to both cyclins and CDKs through a conserved N-terminal domain, blocking their catalytical activity.31
Entering into the cell cycle: Role of CDK4 and CDK6The G1 kinases CDK4 (303 aa, 12q14.1) and CDK6 (326 aa, 7q21.2) share 71% sequence homology and were first described by Meyerson et al. as members of the Cdc2 (also known as CDK1)-related kinase family, along with CDK2 and CDK3.32,49 Despite their phylogenetic distinction from canonical CDKs (CDK1 and CDK2) due to different substrate specificities, CDK4 and CDK6 play crucial roles in cell cycle regulation. Their expression is predominant in the intestinal tract, with CDK4 also present in gynecological tissues such as the ovary and endometrium, and CDK6 in lymphoid tissues, breast, and testis (Human Protein Atlas data).
In normal cells, CDK4 and CDK6 activation requires binding with D-type cyclins (D1, D2, and D3),50 which are multifunctional regulators induced by extracellular mitogenic signals, like the activation of growth factor receptors and integrin-derived adhesion signaling.44
The assembly of cyclin D-CDK4/6 complexes is carefully and temporally regulated during various stages of cell development and differentiation. This regulation varies across cell types, as seen, in hematopoietic, neuronal, and murine embryonic stem cells (mESCs). For instance, cyclin D2 and D3 are highly expressed in most hematopoietic lineages, while cyclin D1 expression is high in embryonic tissue and persist in adult tissue (e.g. in the adult mouse brain). Additionally, cyclin D1 primarily associates with CDK4 in undifferentiated, exponentially growing mESCs but this binding is abolished in G1 arrested mESCs.51,52
CDK4 and CDK6 activation requires phosphorylation by the trimeric CAK complex and dephosphorylation of inhibitory phosphates by CDC25 family members. These kinases enter the cell cycle through phosphorylation at T172 (CDK4) and T177 (CDK6), either as monomers or when complexed with cyclin D and CKIs.53 During the G1 phase, CDC25A removes inhibitory phosphorylation fully activating CDK4 and CDK6. D-type cyclins direct the nuclear translocation of CDK4/6 to phosphorylate retinoblastoma (RB) tumor suppressor family members RB/p105 and RB2/p130, inactivating them.54
CDK4/6 initially phosphorylates RB in G1, with cyclin E-CDK2 driving hyperphosphorylation at the G1/S transition, fully inactivating RB’s nuclear functions.55 Phosphorylation on S807/S811 by CDK4/6, unlike most other sites in RB (i.e. T821, T826), does not seem to cause a structural change nor inhibition of E2F binding, however mutation on these sites prevents the efficient phosphorylation of RB in vivo, sustaining the idea that priming at these sites promotes an intermolecular association to facilitate further phosphorylation.53 Overall, the most accepted model of cell cycle progression in G1 (Fig. 3) suggests that CDK4/6 activity is sufficient to induce RB hyperphosphorylation and E2F activation, while cyclin E/A-CDK2 maintains hyperphosphorylation in the S phase.
Genetic modified mouse models (GEMM) studies have been crucial in understanding the role of CDKs in mammalian development and adult organs physiology. Studies involving CDK4 and CDK6 knockout mice have revealed their redundant and unique non-overlapping functions. Combined ablation of these genes results in specific phenotypes, primarily affecting liver and cardiac tissue.56 CDK4 knockout embryos develop normally, suggesting that CDK4 is not essential for proper mouse development but preferentially affects overall postnatal growth.57 Indeed, they exhibit hypoplasia of various organs, decreased body weight, polyuria, polydipsia, diabetes mellitus, and pancreatic islet degeneration, suggesting a critical role in postnatal proliferation and maintenance of endocrine cells.57 CDK4 depletion also impairs adipocyte function and female fertility, affecting glucose metabolism and the hypothalamic-pituitary axis.58,59 CDK4-deficient MEFs exhibit reduced RB phosphorylation, delaying S-phase entry and extending G1 phase duration.60 On the contrary, MEFs harboring the CDK4 activating mutation R24C, which abrogates p16INK4 inhibitory activity, exhibit decrease doubling times, with a slightly higher percentage of cells in S and G2/M phases, and fail to undergo senescence. Accordingly, CDK4-R24C knock-in mice have increased adipogenic potential, with increased weight of 5-10% compared to control littermates.57,61
CDK6 deletion leads to hematopoietic defects, including thymus and spleen hypoplasia, and reduced megakaryocyte and erythrocyte numbers.56 Combined CDK4/6 knockout results in late embryonic or postnatal lethality due to severe anemia, highlighting their importance in hematopoietic lineage development.56 CDK6 is predominantly localized in the cytoplasm, with a smaller nuclear fraction, indicating additional functions beyond cell cycle progression, such as rapid cell cycle entry post-reactivation in CD8 memory cells and cytoskeletal rearrangement in astrocytes.62
More recently, CDK6 was shown to profoundly reduce thymic cellularity and development, interfering with the proliferative and survival signals activated by NOTCH and AKT pathways.63
Altogether, above mentioned studies revealed that many normal non-transformed mammalian cell types can proliferate without any cyclin D-CDK4/6 activity, suggesting compensatory role of other cyclin-CDKs complexes or pathways.56,64
Although the canonical role of cyclin D and CDK4/6 as essential drivers of cell cycle entry and progression has been firmly established, research carried out over the past two decades provides increasing evidence for additional non canonical functions of these proteins. Among the different activities ascribed to CDK4 and 6 complexes exhaustively reviewed by Hydbring and colleagues, either dependently or independently from their kinase activity,52 we would highlight their important contribution in regulating FOXO transcription factors. Both CDK4 and 6 controls the activity of FOXM1 thereby regulating G2/M transition and cellular senescence.
留言 (0)