Ethics statement
The study received ethics review approval (JSJK2023-B002-02) and provided written informed consent. The murine studies were performed in an animal biosafety level 3 (ABSL3) facility using HEPA-filtered isolators, under controlled conditions of temperature (22–24 °C), a relative humidity of 50–60% and 12-h dark/light cycles. Approval for all animal procedures was obtained from the Institutional Animal Care and Use Committee of the Institute of Laboratory Animal Science, Peking Union Medical College (ILAS, PUMC) under the number BLL22011.
Virus
The SARS-CoV-2 variant Omicron BA.1 (Genbank: OM095411) was discovered at ILAS(PUMC). Other variants of the SARS-CoV-2 virus, including Omicron BA.2.12.1 (GenBank: OP678014), Omicron BA.2 (GenBank: OP678015), Omicron BA.4 (GenBank: OP678017), Omicron BA.5 (GenBank: OP678016), and WH-09 (GenBank: MT093631), were provided by the Guangdong Provincial Center for Disease Control and Prevention (CDC) in China. To confirm the identity of the viral stocks, a plaque-purified viral isolate was expanded as described previously.22 The viral concentration in the supernatant was determined using a standard TCID50 assay method to assess the infectivity of the virus.
Phage library construction
After spending 3-4 weeks recovering in the hospital, peripheral blood mononuclear cells (PBMCs) were extracted from the blood of these resilient patients. The RNA from lymphocytes was carefully isolated and refined using the RNeasy kit from Qiagen, Germany, complying with the manufacturer’s guidelines. Next, cDNA was synthesized by transcribing the total RNA with the Thermo Script reverse transcriptase from Invitrogen, using Oligo-dT as a primer. The heavy and light chain genes were then amplified through polymerase chain reaction (PCR) from the cDNA, and subsequently inserted into the phagemid vector pComb3H obtained from the Scripps Research Institute in the US. Ten clones were randomly selected for sequencing analysis. An anti-SARS-CoV-2 phage antibody library was meticulously created using established primers and techniques.23
Choosing antibodies specific to COVID-19
By utilizing an antibody library, researchers conducted screening with purified SARS-CoV-2 S proteins to isolate potential Fab antibodies. After three rounds of elutriation, the crude Fab antibody preparation underwent testing via an indirect enzyme-linked immunosorbent assay (ELISA), utilizing a 96-well plate coated with purified novel coronavirus S protein at a concentration of 50 ng per well. The second antibody, Horseradish peroxidase (HRP) conjugated anti-human Fab, was employed in the assay. Furthermore, a purification step was conducted using an Anti-Fab affinity chromatography column to isolate human Fab monoclonal antibodies for subsequent characterization and functional analysis. The purity of the human Fab monoclonal antibody was validated through SDS-PAGE analysis.
IgG1 purification methods
The process of expressing and purifying human IgG1 monoclonal antibody involved constructing a complete human IgG-1 antibody by cloning the variable regions of the heavy chain and light chain of a novel SARS-CoV-2-specific antibody into the PIGG vector. This vector, obtained from Scripps Institute, facilitated the expression of both chains of IgG-1. Through transient transfection of human 293 T cells using Lipofectamine 2000, an IgG1 sample was produced from the PIGG vector. Subsequently, the culture medium containing the antibody was purified using affinity chromatography with a recombinant protein AHiTrap column from GE Healthcare. The purity of the IgG1 was then confirmed through SDS-PAGE and ELISA analysis. This method ensured the production of a high-quality and specific human IgG1 monoclonal antibody for potential therapeutic applications.24
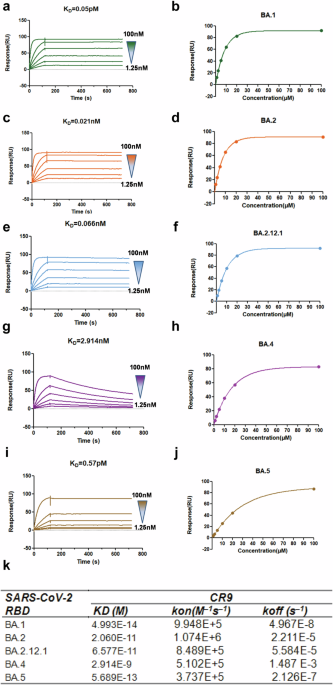
Affinity measurement using Biacore
The study examined the interaction between neutralizing antibodies and the receptor-binding domain (RBD) of SARS-CoV-2 using Surface Plasmon Resonance (SPR) technology. The antibody CR9 was immobilized on a protein A chip and exposed to various concentrations of different SARS-CoV-2 RBD variants. These variants included BA.1, BA.2, BA. 2.12.1, BA.4, and BA.5 at concentrations ranging from 1.25 to 100 nM. By analyzing the binding kinetics and affinity of the antibodies to the RBD variants, researchers aimed to gain insights into the effectiveness of these antibodies in neutralizing the virus. The data obtained was analyzed using BIA Evaluation software to understand the impact of different RBD variants on antibody binding.
Neutralization assay
The determination of the Inhibitory Concentration 50 (IC50) is crucial in the study of antiviral treatments for SARS-CoV-2. This was achieved through observing the cytopathic effects (CPE) of the virus. The process involved serially diluting the antibody two-fold and incubating it with 100 TCID50 SARS-CoV-2 at 37 °C, starting with an initial concentration of 50 μg/ml. Each dilution degree was tested in six replicate wells. Subsequently, the mixture was added to Vero-E6 cells in a 96-well-plate and incubated at 37 °C for 3 days. The experiments carried out in triplicate. The IC50 value was then calculated by analyzing the initial velocity against various concentrations of the combined molecules with a dose-response curve on GraphPad Prism 8.0 software.
Pseudovirus production
A novel approach was taken to create a pseudovirus expressing the protein of the novel coronavirus using the HIV-1 backbone. Through the co-transfection of HEK 293 T cells with specific plasmids, including a packaging plasmid psPAX2, a luciferase reporter gene plasmid pLVX-IRES-ZsGreen, and a plasmid pVAX1encoding the SARS-CoV-2 S proteins, a pseudovirus carrying the novel coronavirus protein and expressing luciferase was successfully constructed. This innovative method opens up new possibilities for studying the novel coronavirus and its interactions with host cells.
After incubating cells with the transfection mixture for 24 h, fresh DMEM medium with 10% FBS was added to each Petri dish. The culture medium was then replaced with fresh DMEM medium after overnight incubation. 48 hs post-transfection, the culture supernatant was collected and stored at -80°C for later use.
Pseudovirus neutralization assay
Pseudovirus neutralization assay measures the ability of antibodies to neutralize pseudoviruses, which are engineered viruses that mimic the structure of real viruses but do not cause illness. This assay is a critical tool in vaccine development and evaluating the efficacy of antibody-based treatments. It allows researchers to assess how well antibodies can prevent viral entry into cells, providing valuable insights into the potential effectiveness of immune responses. Monoclonal antibodies were diluted and tested against pseudovirus in pairs to determine effectiveness. Study shows efficacy of neutralization assay with human ACE2-overexpressed HEK 293 T cells. Pseudoviruses were introduced to 293T-hACE2 cells and incubated at 37 °C for an hour. Subsequently, 293T-hACE2 cells were added to each well and plates were further incubated. After two days, luciferase activity was assessed to determine the effectiveness of the treatment. The IC50 value was determined by analyzing the initial velocity and varying concentrations of the compounds in GraphPad Prism 8.0 software. This process allowed for a precise measurement of the inhibitory concentration required to combat the pseudovirus infection in the cells.
Animal experiments
The study conducted by the Institute of Laboratory Animal Science, Peking Union Medical College utilized specific pathogen-free (SPF) hACE2 mice aged 6-8 weeks to investigate the prophylactic and therapeutic efficacy of purified antibodies against SARS-CoV-2 variants BA.2.12.1 and BA.5. The mice were randomly divided into eight groups, with each group consisting of five mice. For the prophylactic assessment, the mice were injected with 5 mg/kg of the purified antibody 24 h before being intranasally challenged with either SARS-CoV-2 variant at a dosage of 105 TCID50 in 50 μL of phosphate-buffered saline (PBS). As for the therapeutic evaluation, mice were first infected with 105 TCID50/mL of the SARS-CoV-2 variants and then treated with either 10 or 20 mg/kg of the purified monoclonal antibody after 2 h. Observations were made daily post-infection, recording changes in body weight, clinical symptoms, and responsiveness to external stimuli. The mice were sacrificed at 4 days post-infection, and their lungs were collected for viral load analysis and pathological examination. This research provides valuable insights into the potential effectiveness of purified antibodies in preventing and treating SARS-CoV-2 infections in mice.
Quantitative RT-PCR for mRNA
Total RNA was extracted using a standard method and then converted to cDNA through RT-PCR.25 Subsequently, qRT-PCR was conducted with a specific cycling protocol and primers. The cycling protocol included initial steps at 50 °C for 2 min and 95 °C for 2 min, followed by 40 cycles at 95 °C for 15 seconds and 60 °C for 30 seconds. Final incubation steps were carried out at 95 °C for 15 seconds, 60 °C for 1 minute, and 95 °C for 45 seconds. To detect SARS-CoV-2, specific primers were utilized.
Protein expression and purification
The plasmid encoding the full spike protein (S protein) of SARS-CoV-2 was utilized as a template to create a variant known as BA.5, which contains multiple mutations. To achieve this, overlapping PCR techniques were employed. The resulting S gene of BA.5 was then inserted into a pCAGGS vector, along with a T4 fibritin trimerization motif, an HRV3C protease site, and a Twin-Strep-tag at the C-terminus, in accordance with previously established methods. Additionally, the receptor-binding domain (RBD) of BA.5 was derived from the S gene using similar PCR techniques and cloned into a pCAGGS vector with a 10His tag. Subsequently, the expression vector containing the modified genes was transiently transfected into HEK293F cells grown in suspension under specific conditions. Following a 72-hour incubation period, the supernatant containing the target proteins was collected, concentrated, and subjected to buffer exchange. The S protein was isolated using chromatography with streptavid in resin and further purified through size exclusion chromatography. Similarly, the RBD protein was purified using nickel-charged resin and size exclusion chromatography, all conducted in buffer solutions of appropriate pH and salt concentration. This meticulous process yielded highly purified BA.5 spike protein and RBD proteins, ready for further characterization and potential applications in research and development. The detailed methodology ensures the integrity and quality of the proteins for future studies exploring the impact of the identified mutations on viral infectivity and immune response.
Cryo-EM sample collection, data acquisition, and structure determination
Cryo-electron microscopy (Cryo-EM) samples were prepared by combining the BA.5 S trimer with CR9 antibody Fab fragments in a specific ratio and chilling them on ice to form the S-Fab complex. This complex was then carefully placed onto grids that had been treated to enhance adhesion. By applying a brief 6-second blotting at full humidity, the grids were plunged into liquid ethane using a Vitrobot system, ensuring the samples were perfectly preserved. High-quality movies of the complex were captured with a K3 Summit direct detector, with precise defocusing between 1.5–2.7 μm to capture all details. The data acquisition process was automated using Serial EM at a magnification of 22,500x, resulting in an impressive final pixel size of 1.07 Å. This meticulous process allowed for the detailed analysis of the S protein and Fab complex at an atomic level.
In order to enhance the precision of the binding interface between SARS-CoV-2 BA.5 RBD and CR9, researchers successfully determined the structure of BA.5 RBD-CR9. Utilizing the Fab SN1600, a class IV tool known for boosting molecular weight for improved contrast, a cryo-EM sample complex CR9-BA.5 RBD-SN1600 was prepared at a molar ratio of 1:1.2:1.2, resembling the CR9-Spike complex. Data collection took place at 300 kV using an FEI Titan Krios microscope. Movies were recorded with a Gatan K2 Summit direct electron detector by Serial EM, featuring 32 frames lasting 0.2 seconds each and a total dose of 60 e/A°-2s-1. The defocus range fell between 1.5–2.4 μm. The calibrated magnification was set at 22,500×, resulting in a final pixel size of 1.04 A°.
Totally 4541micrographs of the Spike-CR9 complex and 3943 micrographs of RBD-CR9-SN1600were collected. CryoSPARC was used to correct beam-induced motion and average the frames. The defocus value of each micrograph was then estimated using the patch CTF estimation. Then, 2,244,585 Particles of Spike-CR9 complex and 1,118,175 particles of RBD-CR9-SN1600 complex were auto picked and extracted for 2D classification. Next, 364,898good-quality particles of Spike-CR9 complex were primarily selected for Ab-intio reconstruction into 5 classes to generate initial references, and 613,672 particles of RBD-CR9-SN1600 complex were reconstructed in Ab-initio reconstruction in 4classes. Then, heterogenous refinement was performed using the particles and references from Ab-intio reconstruction for both Spike-CR9 and RBD-CR9-SN1600 complexes. At last, 2 classes of Spike-CR9 from heterogenous refinement were selected for homogenous refinement to obtain the final cryo-EM density. One class of RBD-CR9-SN1600 were selected for homogenous refinement and non-uniform refinement for the final cryo-EM density.
Resolutions were assessed using the standard Fourier shell correction at a threshold of 0.143 with ResMap. Data processing steps are detailed in Supplementary Fig. S1 for transparency and reproducibility in evaluating resolution quality.
Model fitting and refinement
The generation of atom models for the Spike-CR9 complex involved fitting the apo BA.5 spike (PDB:7XNQ) and Fab (PDB:7E5Y) chains into cryo-EM density using Chimera. The resulting structure underwent manual adjustments in Coot, considering protein sequences and density, followed by real-space refinement with Phenix. Similarly, the RBD-CR9-SN1600 complex was derived from the Spike-CR9 complex using Chimera and subsequently refined with Phenix. These refinement procedures are outlined in Supplementary Fig. S1.
Statistical analysis
Statistical analysis was conducted using GraphPad Prism 8.0 software. Group comparisons were made with a two-tailed, unpaired Student’s t-test. Significance levels were set at *p < 0.05 and **p < 0.01 to determine statistical significance.



留言 (0)