
記住我

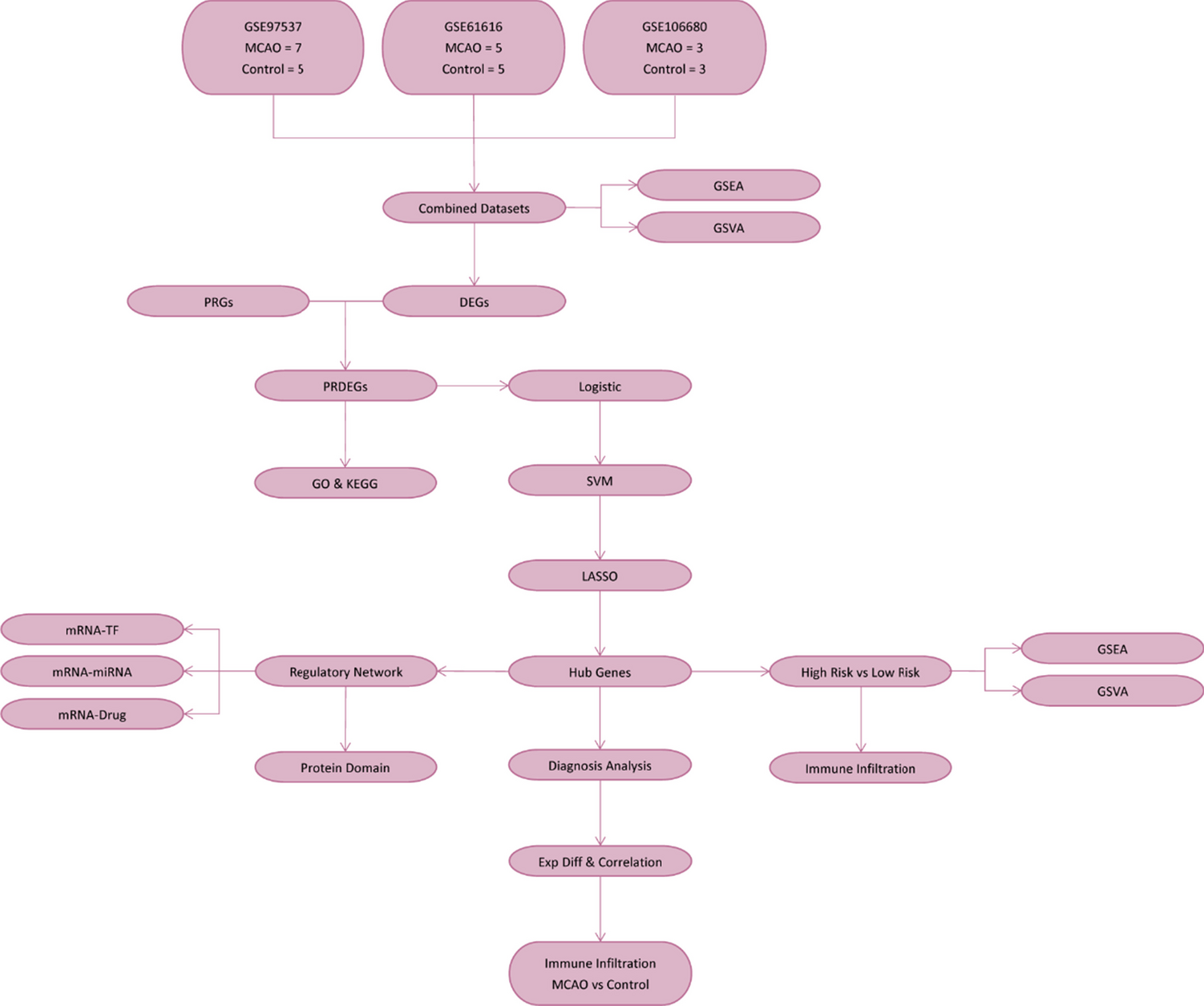


We obtained MCAO datasets (GSE97537, GSE61616, GSE106680, n = 28) from GEO databases. PRGs sourced from GeneCards and the MSigDB underwent differential expression analysis, followed by GO and KEGG functional assessments. Functional enrichment differences between high-risk and low-risk MCAO groups were calculated using GSEA and GSVA. LASSO regression identified and validated PRDEGs for constructing the MCAO diagnostic model. High- and low-risk MCAO groups underwent expression and immune cell correlation analysis with PRhubGs. A regulatory network between PRhubGs and miRNA was established, and protein domain prediction was conducted (Fig. 1).
Fig. 1
Flow chart for the comprehensive analysis of PRDEGs. MCAO, middle cerebral artery occlusion; DEGs, differentially expressed genes; PRGs, pyroptosis-related genes; GSEA, gene set enrichment analysis; GSVA, gene det variation analysis; GO, Gene Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes; PRDEGs, pyroptosis-related differentially expressed genes; SVM, support vector machine; LASSO, least absolute shrinkage and selection operator; TF, transcription factor
Integration of the MCAO DatasetsThe MCAO datasets were involved in initial batch effect removal and formed a unified GEO dataset termed “combined datasets.” The PCA graphs revealed a significant reduction in batch effects among the samples within the MCAO dataset following the batch effect removal procedure (Fig. 2A–D).
Fig. 2
Batch effects removal of GSE97537, GSE61616, and GSE106680. A, B Boxplots depicting the distribution of the Combined Datasets before (A) and after (B) batch effects removal. C, D Principal component analysis (PCA) plots illustrating the datasets’ distribution before (C) and after (D) batch effects removal. Colors represent different MCAO datasets: purple indicates GSE106680, green indicates GSE61616, and yellow indicates GSE97537
Analysis of DEGs Associated with Cell PyroptosisThe combined datasets were stratified into MCAO and control groups. Differential expression analysis unveiled a total of 675 DEGs, of which 507 were upregulated and 168 were downregulated (Fig. 3A). We intersected all DEGs and PRGs and determined a total of 25 PRDEGs (Fig. 3B), namely PLIN2, MCM2, HSPB1, CDT1, MCM3, CKS2, CKLF, MCM6, MCM5, MELK, CDCA3, CDCA7, SDF2L1, CCNA2, UHRF1, PBK, TOP2A, LOX, PRC1, AKR1B10, WISP2, FAM111A, AURKB, BUB1B, and TTK.
Fig. 3
Differential gene expression analysis for Combined Datasets. A Volcano plot showing the analysis of differentially expressed genes between the MCAO and Control groups in the integrated GEO datasets (Combined Datasets). B Venn diagram displaying the overlap between DEGs and PRGs in the integrated GEO datasets (Combined Datasets). C Correlation heatmap of PRDEGs in the integrated GEO datasets (Combined Datasets). D Chromosomal localization plot of PRDEGs. Colors represent Control (green) and MCAO (yellow) groups. MCAO: middle cerebral artery occlusion; DEGs, differentially expressed genes; PRGs, pyroptosis-related genes; PRDEGs, pyroptosis-related differentially expressed genes
Subsequently, the 25 PRDEGs among different sample groups in the combined datasets showed differential expressions (Fig. 3C). The 25 PRDEGs were distributed on each chromosome (Fig. 3D). Notably, most of the PRDEGs were located on chromosome 9, including CKS2, MELK, and PLIN2.
GO and KEGG Enrichment AnalysesGO and KEGG enrichment analyses were conducted to elucidate the biological relevance of the 25 PRDEGs in the context of MCAO (Table 2 and Fig. 4A). The PRDEGs were predominantly enriched in biological processes (BPs) such as double-strand break repair via break-induced replication, DNA replication initiation, negative regulation of chromosome organization, DNA unwinding involved in DNA replication, and DNA-templated DNA replication. Regarding cellular components (CCs), enrichment was observed in chromosomal regions, CMG complex, DNA replication preinitiation complex, chromosome centromeric regions, and nuclear chromosomes. In terms of molecular functions (MFs), the PRDEGs were associated with single-stranded DNA helicase activity, ATP-dependent activity acting on DNA, single-stranded DNA binding, 3′−5′ DNA helicase activity, and DNA helicase activity. Additionally, significant enrichment was identified in biological pathways such as DNA replication and cell cycle. Furthermore, network diagrams were generated to depict the relationships among BP, CC, MF, and pathways (Fig. 4B–E).
Table 2 Results of GO and KEGG enrichment analysis for PRDEGsFig. 4
GO and KEGG enrichment analysis for PRDEGs. A Bar charts illustrating Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis results for pyroptosis-related differentially expressed genes (PRDEGs), categorized into biological process (BP), cellular component (CC), molecular function (MF), and pathway. The x-axis represents GO terms and KEGG terms. B–E Network diagrams presenting the results of GO and KEGG enrichment analysis for PRDEGs, showcasing BP (B), CC (C), MF (D), and KEGG pathways (E). Pink nodes represent entries, blue nodes represent molecules, and lines denote relationships between entries and molecules
Finally, the enriched KEGG pathways, particularly DNA replication (Fig. 5A) and cell cycle (Fig. 5B), were visualized using the Pathview R package.
Fig. 5
KEGG pathway visualization. A, B Pathway visualization of Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis results for pyroptosis-related differentially expressed genes (PRDEGs), highlighting DNA replication (A) and cell cycle (B)
GSEA of the Integrated GEO DatasetGSEA was employed to elucidate the impact of gene expression levels on MCAO within the integrated GEO dataset. The analysis was conducted to explore the connection between the expression of all genes in the integrated GEO dataset and the biological processes involved, cellular components affected, and molecular functions exhibited (Fig. 6A). Detailed results are presented in Table 3. The GSEA revealed significant enrichment of all genes in the integrated GEO dataset in biologically relevant functions and signaling pathways, notably in the IL18 signaling pathway (Fig. 6B), inflammatory response pathway (Fig. 6C), il1 and megakaryocytes in obesity (Fig. 6D), and oxidative damage response (Fig. 6E).
Fig. 6
GSEA for combined datasets. A Gene set enrichment analysis (GSEA) mountain plots depicting the enrichment of four biological functions in the integrated GEO datasets (combined datasets). B–E GSEA showing significant enrichment of middle cerebral artery occlusion (MCAO) in IL18 signaling pathway (B), inflammatory response pathway (C), il1 and megakaryocytes in obesity (D), and oxidative damage response (E)
Table 3 Results of GSEA for combined datasetsGSVA of the Integrated GEO DatasetGSVA was conducted to investigate the differential enrichment of the c2.all.v7.5.1.symbols.gmt gene set between the MCAO and control groups within the combined dataset (Table 4 and Fig. 7A). The GSVA results indicated that 12 pathways exhibited statistically significant differences (p value < 0.05) between the MCAO and control groups. These pathways include macrophage markers; ZERBINI response to SULINDAC DN, CLEC7A inflammasome pathway, TESAR ALK targets human ES 4D DN, TESAR ALK targets human ES 5D DN, RUNX3 regulates immune response and cell migration, CD22 mediates BCR regulation, metal sequestration by antimicrobial proteins, GALI TP53 targets apoptotic UP, RUNX1 regulates transcription of genes, SMIRNOV circulating endotheliocytes in cancer DN, and activation of C3 and C5. The GSVA scores of the 12 pathways also showed differences between the MCAO and control groups (Fig. 7B).
Table 4 Results of GSVA for combined datasetsFig. 7
GSVA for combined datasets. A, B Gene set variation analysis (GSVA) results in boxplots (A) and heatmap (B) for the MCAO and control groups in the integrated GEO datasets. Green represents the Control group and yellow the MCAO group. ***p value < 0.001, highly statistically significant
MCAO Diagnostic ModelA logistic regression model was constructed based on these genes to ascertain the diagnostic value of the 25 PRDEGs in MCAO (Fig. 8A). We identified 14 statistically significant PRDEGs (p value < 0.05), namely AURKB, BUB1B, CCNA2, CDCA3, CDCA7, CKLF, FAM111A, MELK, PBK, PRC1, SDF2L1, TOP2A, TTK, and WISP2.
Fig. 8
Diagnostic model of MCAO. A Forest plot illustrating 25 PRDEGs included in the logistic regression model for the MCAO diagnostic model. B, C Visualization of the minimum gene count (B) and maximum accuracy gene count (C) using the SVM algorithm. D, E Diagram (D) and variable trajectory plot (E) of the LASSO regression model’s diagnostic model. F Nomogram depicting pyroptosis-related hub genes in the diagnostic model for MCAO based on the integrated GEO datasets. MCAO, middle cerebral artery occlusion; SVM, support vector machine; LASSO, least absolute shrinkage and selection operator; PRDEGs, pyroptosis-related differentially expressed genes
Subsequently, an SVM model was developed using the 14 PRDEGs and SVM algorithm, revealing optimal performance with a gene count of 4 (Fig. 8B, 7). These 4 PRDEGs—WISP2, MELK, SDF2L1, and AURKB—were key contributors to the SVM model. A LASSO regression analysis based on these genes yielded the final MCAO diagnostic model (Fig. 8D–E).
A nomogram (Fig. 8F) was constructed based on the cell death-related hub genes (hub genes) to validate the diagnostic model’s utility further. Results demonstrated that the expression level of WISP2 significantly contributed to the diagnostic model’s efficacy, surpassing other variables, while MELK exhibited lower utility compared with other variables.
Validation of the MCAO Diagnostic ModelROC curves were generated using the expression levels of the cell death-related hub genes (hub genes) WISP2, MELK, SDF2L1, and AURKB in the combined datasets to validate the diagnostic model for MCAO. The ROC curves indicated high accuracy (AUC > 0.9) in distinguishing different groups (Fig. 9A–D).
Fig. 9
Diagnostic analysis of MCAO. A–D ROC curves for pyroptosis-related hub genes in the integrated GEO datasets. E Violin plot displaying the PScore in the MCAO and Control groups in the integrated GEO datasets. F ROC curve for the PScore in the integrated GEO datasets. G, H Calibration curve (G) and DCA plot (H) for the MCAO diagnostic model based on the PScore in the integrated GEO datasets. MCAO, middle cerebral artery occlusion; ROC, receiver operating characteristic; PScore, pyroptosis score; DCA, decision curve analysis
Utilizing the ssGSEA algorithm, PScores were calculated based on the expression of the four hub genes across all samples in the combined datasets. The PScore between the MCAO and control groups showed significant variations (p value < 0.001) (Fig. 9E). Furthermore, ROC curves based on PScore confirmed their high accuracy (AUC > 0.9) in distinguishing different groups (Fig. 9F).
The accuracy and distinguishing power of the MCAO diagnostic model were evaluated through calibration analysis, presenting a calibration curve that slightly deviated from the ideal diagonal line but demonstrated a close fit (Fig. 9G). Additionally, DCA based on PScore from the combined datasets assessed the clinical utility of the MCAO diagnostic model, revealing a stable net benefit within a certain range and overall favorable performance (Fig. 9H).
GSEA of the High- and Low-Risk GroupsThe MCAO samples were stratified into high- and low-risk groups based on their RiskScores, calculated using the formula:
$$RiskScore=WISP2\times 1.6643+MELK\times 0.4164+SDF2L1\times 1.8724+AURKB\times 0.8093.$$
Differential expression analysis identified 18 DEGs in MCAO samples (adjusted p value < 0.05), of which 12 were upregulated (logFC > 1.00, adjusted p value < 0.05), and 6 were downregulated (logFC ≤ 1.00, adjusted p value < 0.05) (Fig. 10A–B). GSEA was performed to understand the impact of gene expression in MCAO samples on the condition. Notably, all genes in MCAO samples were significantly enriched in processes such as apoptosis (Fig. 10D), il1 and megakaryocytes in obesity (Fig. 10E), IL18 signaling pathway (Fig. 10F), and photodynamic therapy-induced Nfkb survival signaling (Fig. 10C, 10, Table 5).
Fig. 10
GSEA for risk group. A Volcano plot illustrating differentially expressed genes between the high- and low-risk groups in MCAO samples. B Correlation heatmap of DEGs in the MCAO samples. C GSEA mountain plots showing significant enrichment of MCAO in apoptosis (D), il1, and megakaryocytes in obesity €, IL18 signaling pathway (F), and photodynamic therapy-induced Nfkb survival signaling (G). MCAO, middle cerebral artery occlusion; GSEA, gene set enrichment analysis; DEGs, differentially expressed genes
Table 5 Results of GSEA for risk groupGSVA of the High- and Low-Risk GroupsTo explore the differences in the c2.all.v7.5.1.symbols.gmt gene set between the high- and low-risk groups in MCAO samples, GSVA was conducted on all genes from MCAO samples (Table 6).
Table 6 Results of GSVA for risk groupGSVA revealed five pathways with significant differences (p-value < 0.05) between the two groups, namely, attachment and entry, MYLLYKANGAS amplification hot spot 21, LAU apoptosis CDKN2A DN, RUNX1 regulates transcription of genes, and NECTIN NECL trans heterodimerization (Fig. 11A, 11).
Fig. 11
GSVA for risk group. A, B GSVA results in boxplots (A) and heatmap (B) for the high- and low-risk groups in MCAO samples. Green represents the low-risk group, and yellow is the high-risk group. **p value < 0.01, highly statistically significant; ***p value < 0.001, extremely statistically significant
Correlation and Expression Analysis of Cell Death-Related Hub GenesThe four hub genes WISP2, MELK, SDF2L1, and AURKB showed significant differential expression between the MCAO and control groups (p value < 0.001, Fig. 12A) and between the high- and low-risk groups (p value < 0.05). Specifically, WISP2 showed highly significant expression differences (p value < 0.001), while MELK and SDF2L1 showed substantial statistical significance (p value < 0.01). The four hub genes exhibited a significant positive correlation among the cell death-related hub genes in both the integrated GEO dataset and MCAO samples (Fig. 12C–D).
Fig. 12
Correlation and expression difference analysis for hub genes. A Violin plots illustrating pyroptosis-related hub genes in the integrated GEO datasets. B Violin plots showing pyroptosis-related hub genes in MCAO samples. C Correlation heatmap of pyroptosis-related hub genes in the integrated GEO datasets. D Correlation heatmap of pyroptosis-related hub genes in MCAO samples. Green represents the control group, and yellow is the MCAO group. **p value < 0.01, highly statistically significant; ***p value < 0.001, extremely statistically significant. The color intensity indicates the correlation coefficient (r value); wider bands represent larger absolute values of the correlation coefficient
Integrated GEO Dataset Immuno-Infiltration AnalysisThe CIBERSORT algorithm was applied to compute the correlation between 22 immune cell types and the samples from the MCAO and control groups. Immuno-infiltration analysis revealed the proportions of immune cells in the Integrated GEO Dataset (Fig. 13A), demonstrating positive enrichment scores for 19 immune cell types (Fig. 13B). A total of 12 immune cell types showed significant expression differences (p value < 0.05) between the MCAO and control groups, namely memory B cells, CD8 T cells, CD4 naive T cells, regulatory T cells (Tregs), monocytes, M0 macrophages, M1 macrophages, resting dendritic cells, resting mast cells, activated mast cells, eosinophils, and neutrophils (Fig. 13B). Notably, positive correlations were observed between CD4 naive T cells and memory B cells (r value = 0.68) and negative correlations between resting dendritic cells and memory B cells, as well as neutrophils and Tregs (r value = − 0.65) (Fig. 13C). Furthermore, a significant positive correlation was observed between the cell death-related hub gene WISP2 and the immune cell type dendritic cells resting, and a notable negative correlation was observed between MELK and the immune cell type macrophages M1 (Fig. 13D).
Fig. 13
Combined datasets immune infiltration analysis using CIBERSORT Algorithm. A, B Bar charts depicting the proportions of immune cells (A) and boxplots for group comparison (B) in the integrated GEO datasets. C Correlation heatmap of immune cell infiltration abundance in the integrated GEO datasets. D Correlation plot of pyroptosis-related hub genes in the integrated GEO datasets with immune cell infiltration abundance. Green represents the control group, and yellow is the MCAO group. Blue indicates a negative correlation, and pink indicates a positive correlation; The correlation coefficient (r value) between 0.3 and 0.5 is considered weak, and between 0.5 and 0.8 is moderate. MCAO, middle cerebral artery occlusion
Immunoinfiltration Analysis of the High- and Low-Risk GroupsThe ssGSEA algorithm was applied to calculate the immune infiltration abundance of 28 immune cell types in the high- and low-risk groups (p value < 0.05; Fig. 14A). Twelve immune cell types showed significant differences (p value < 0.05), namely activated CD4 T cells, central memory CD4 T cells, effector memory CD4 T cells, effector memory CD8 T cells, gamma delta T cells, regulatory T cells, type 1 T helper cells, activated dendritic cells, immature dendritic cells, myeloid-derived suppressor cells (MDSCs), natural killer cells, and plasmacytoid dendritic cells. Correlation heatmaps demonstrated the relationships among the infiltration abundances of the 12 immune cell types in different groups (Fig. 14B–C). Positive correlations were observed in both the high- and low-risk groups for all immune cell types except the effector memory CD8 T cells, with the low-risk group showing more negative correlations.
Fig. 14
Risk group immune infiltration analysis by ssGSEA algorithm. A Boxplots illustrating the group comparison of immune cells in the MCAO disease subtypes. B, C Correlation analysis results for immune cell infiltration abundance in the high-risk (B) and low-risk (C) groups in MCAO. D–E Correlation plots depicting the relationship between immune cell infiltration abundance and pyroptosis-related hub genes in the high-risk (D) and low-risk (E) groups in MCAO. ssGSEA, single-sample gene-set enrichment analysis; MCAO, middle cerebral artery occlusion. Green represents the low-risk group, and yellow is the high-risk group. *p value < 0.05, statistically significant; **p value < 0.01, highly statistically significant; ***p value < 0.001, extremely statistically significant. The color intensity indicates the correlation coefficient (correlation); pink represents a positive correlation, and blue is a negative correlation
Finally, correlation dot plots showcased the associations between cell death-related hub genes and the immune infiltration abundances in the high- and low-risk groups of the MCAO dataset (Fig. 14D–E). In the high-risk group, a significant positive correlation was observed between the immune cell-activated dendritic cell and the hub gene SDF2L1. Conversely, in the low-risk group, a notable positive correlation was observed between the immune cell Natural killer cell and the hub gene SDF2L1.
Regulatory Network Construction and Protein Domain PredictionWe initially obtained TF binding to cell death-related hub genes from the ChIPBase database, forming the mRNA-TF regulatory network to elucidate the regulatory landscape (Fig. 15A). The network comprised two cell death-related hub genes and 53 TFs. Utilizing the TarBase database, we identified miRNAs associated with cell death-related hub genes, constructing the mRNA-miRNA regulatory network. Visualization of this network through Cytoscape (Fig. 15B) revealed four hub genes involving 41 miRNAs. Exploration of the CTD database facilitated the acquisition of drugs linked to cell death-related hub genes, leading to the construction of the mRNA-drug regulatory network. Cytoscape visualization (Fig. 15C) showcased two hub genes and 18 associated drugs. To gain insights into the protein architecture of the four-cell death-related hub genes, including WISP2, MELK, SDF2L1, and AURKB, we leveraged the SMART database for protein domain prediction (Fig. 15D-G). Notably, WISP2 contained the domains IB, VWC, and TSP1; MELK contained the S_TKc domain; SDF2L1 contained the MIR domain; AURKB contained the S_TKc domain.
Fig. 15
Regulatory network and protein domains of hub genes. A mRNA-TF regulatory network of pyroptosis-related hub genes. B mRNA-miRNA regulatory network of pyroptosis-related hub genes. C mRNA-drug regulatory network of pyroptosis-related hub genes. D–G Predicted protein domains of pyroptosis-related hub genes WISP2 (D), MELK (E), SDF2L1 (F), and AURKB (G). TF, transcription factor. Blue nodes represent TFs, green nodes represent miRNAs, and purple nodes represent drugs
Drug Susceptibility AnalysisTo explore the appropriate drug treatment strategies for patients with cerebral ischemia–reperfusion injury (MCAO), we used drug susceptibility data from the GDSC database as a training set to predict the sensitivity of the cerebral ischemia–reperfusion injury (MCAO) group in the integrated GEO datasets to common anticancer drugs. We then retained the top 20 drugs that differed significantly between the high-risk and low-risk groups: Docetaxel, Doxorubicin, Midostaurin, X17. AAG, WZ.1.84, Parthenolide, Etoposide, Mitomycin.C, Vinorelbine, Pazopanib, CEP.701, Cyclopamine, Bicalutamide, GSK.650394, Thapsigargin, Bexarotene, Bleomycin, AUY922, Tipifarnib and Camptothecin, and the results were presented (Fig. 16A–T). We found that among the 20 drugs with significant differences, drug sensitivity was generally lower in the high-risk group than in the low-risk group (Fig. 16A–T).
Fig. 16
Results of drug susceptibility analysis. Based on the GDSC database, the integrated GEO dataset was used to evaluate the treatment of Docetaxel (A), Doxorubicin (B), Midostaurin (C), X17 between the high-risk group and the low-risk group in the cerebral ischemia–reperfusion injury (MCAO) group. AAG(D), WZ.1.84(E), parthenolide(F), etoposide(G), mitomycin.C(H), vinorelbine(I), pazopanib(J), CEP.701(K), cyclopamine(L), bicalutamide(M), GSK.650394(N), thapsigargin(O). The sensitivity analysis results of bexarotene (P), bleomycin (Q), AUY922 (R), tipifarnib (S), and camptothecin (T) were grouped and compared
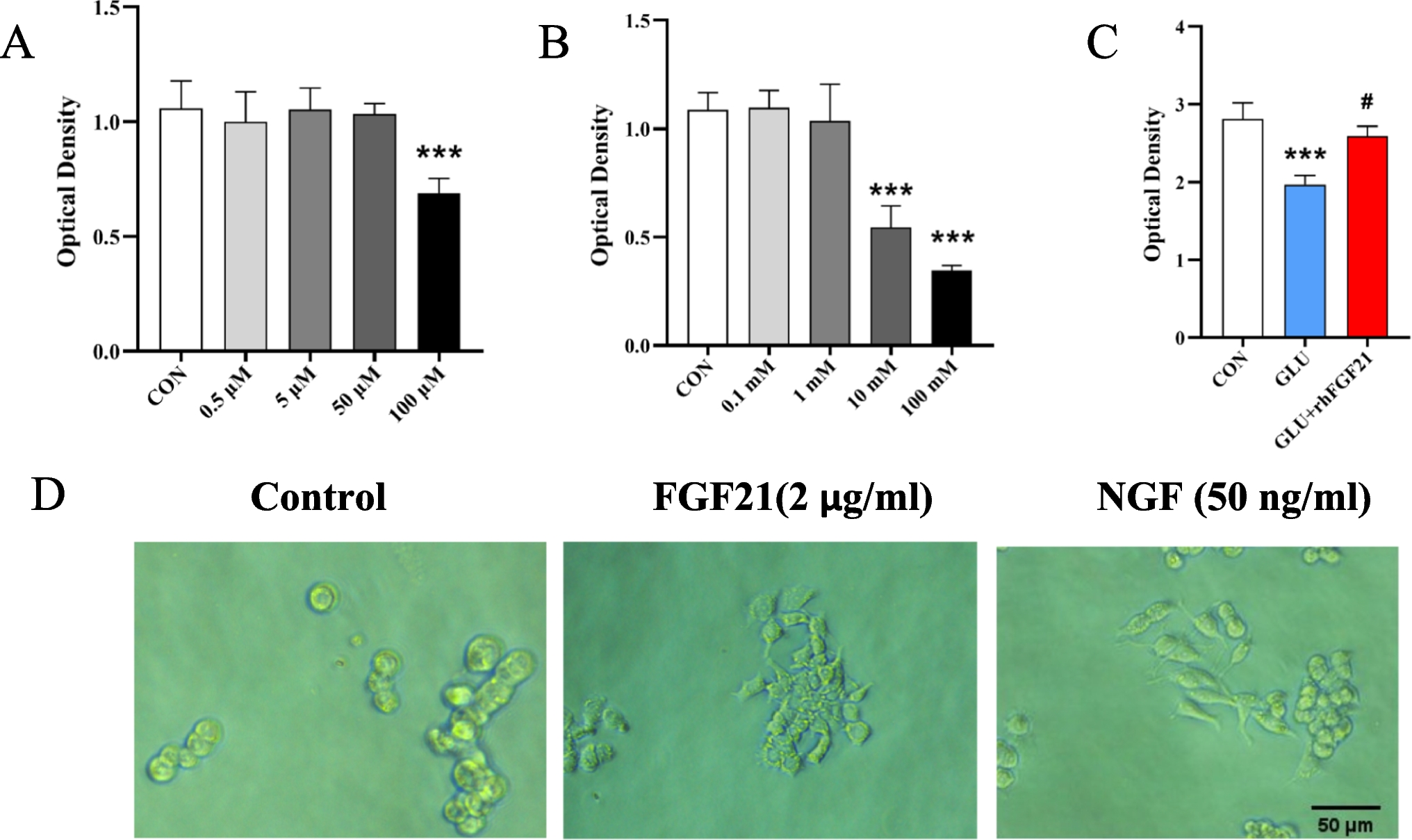
Validation of WISP2, MELK, SDF2L1, and AURK Using qRT-PCRThe MCAO rat model was constructed to simulate IS. The expression of four characteristic genes in the brain-damaged areas of MCAO and sham rats was verified using RTqPCR. The expression of WISP2 (Fig. 17A), MELK (Fig. 17B), and SDF2L1(Fig. 17C) in MCAO was significantly higher than that in sham samples; however, no significant difference in AURKB gene expression was observed (Fig. 17D).
Fig. 17
The expression levels of WISP2 (A), MELK (B), SDF2L1 (C), and AURKB (D) in ischemic stroke brain tissue were measured by RT-qPCR. *p < 0.05, **p < 0.01, ***p < 0.001
留言 (0)