Mice
Female BALB/c (strain no. 000651) and CD45.1-BALB/c (strain no. 006584) mice were obtained from The Jackson Laboratory. All mice were between 6 and 8 weeks of age at the start of the experiment. The numbers of mice used are indicated in the figure legends. Mice were housed in specific pathogen-free animal facilities under a 12 h light–dark cycle at the University of Connecticut Health Center. The temperature in the housing facilities is kept at a maximum of 24 °C and a minimum of 21 °C. Humidity in the room is maintained at a maximum of 61% and a minimum of 25%. Mice were fed diet nos. 2918 and 2919 (Inotiv). All experiments conducted were part of animal protocols approved by the Animal Care and Use Committee of the University of Connecticut School of Medicine (AP-2000633-0325).
Tumor growth assay
Meth A cells that have been in our laboratory since 1986 were originally obtained from Lloyd J. Old. They were passaged in ascites and were free from Mycoplasma contamination. The MC38-FABF tumor cell line was provided by A. B. Frey at the New York University Langone Medical Center in 2010. Cells were screened for Mycoplasma and tested negative.
Meth A cells were grown in the ascites of mice for 4 days before injection (95,000 live cells injected intradermally into the right flank of mice). MC38-FABF cells were grown in culture as described elsewhere17. Tumors on mice were measured twice weekly with calipers for about 6 weeks. The maximum tumor size or burden permitted by our ethics committee is a diameter of 2 cm per 4.2 cm3volume. This limit was not violated. Mice with tumors exceeding a 15-mm diameter were euthanized. Tumor volumes were calculated using the equation 0.4 × l × w2 where l represents the length and w represents the width of the tumor. The tumor control index was calculated as described in ref. 54, using average tumor diameter measurements.
In vivo immune checkpoint blockade
Isotype-treated mice were injected twice weekly (intraperitoneally) with 65 µg of mouse IgG2b (clone MPC-11) and weekly with 30 µg of rat IgG2a (clone 2A3) antibody. Anti-PD-1-treated mice were injected twice weekly (intraperitoneally) with 65 µg of mouse IgG2b and weekly with 30 µg of αPD-1 (clone RPMI-14) antibody. Anti-CTLA-4-treated mice were injected twice weekly (intraperitoneally) with 65 µg αCTLA-4 (clone 9D9) and weekly with 30 µg of Rat IgG2a antibody.
Isolation of CD8+ T cells from tumors and LNs
Tumors were excised from mice, using scalpels, 28 days after tumor challenge and digested using the Tumor Dissociation Kit, mouse on a gentleMACS Octo Dissociator (Miltenyi Biotec). The digested tumor solution was then washed twice with cold PBS and subjected to density-based separation of lymphocytes using Lympholite M (Cedarlane Laboratories). LNs were excised and subjected to EasySep Mouse CD8+ T cell Isolation Kit (STEMCELL Technologies).
Modified tetramer decay assay
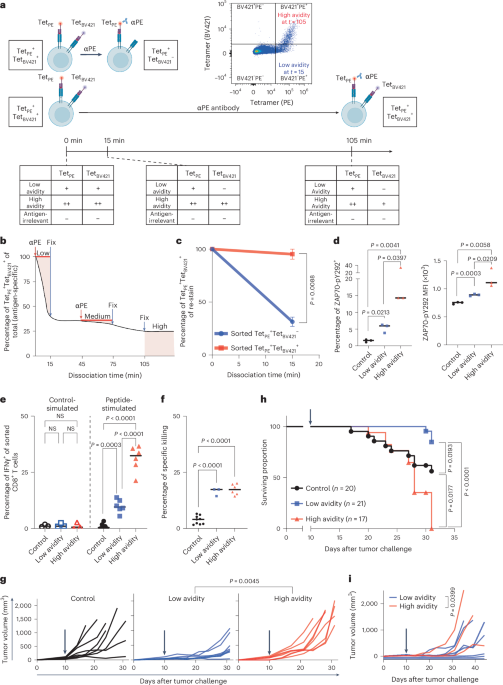
CD8+ T cells, extracted as described in ‘Isolation of CD8+ T cells from tumors and LNs’, were then subjected to Fc blocking with mouse IgG isotype antibody (1:50 dilution, Invitrogen) for 20 min on ice. Cells were washed and resuspended in tetramer staining solution containing 1% v/v tetramers (MBL Technologies) loaded with the same antigen labeled with either BV421 or PE at a concentration of 10 million cells per ml for 30 min on ice. The tetramer staining solution was then washed off and cells were stained with a surface antibody staining solution for 20 min on ice. We observed that inclusion of an anti-PE antibody during tetramer decay prevented the dissociation of TetPE over 120 min; thus, the tetramer was stabilized (Extended Data Fig. 1c, right and Extended Data Fig. 1g). In contrast, the nonstabilized TetBV421 dissociated normally (Extended Data Fig. 1d). Stabilized binding of TetPE served as the reference point against which decay of TetBV421 was compared. The addition of a stabilizing antibody was a critical innovation in this assay: in its absence, low-avidity and medium-avidity cells are indistinguishable from antigen-nonspecific T cells because they are also tetramer-negative.
Therefore, cells were split into three samples and subjected to tetramer decay at room temperature in anti-H2-Dd (clone 34-5-8S, 20 µg ml−1) antibody for either 15 (low avidity), 75 (medium avidity) or 105 (high avidity) minutes. Anti-PE (clone PE0001, 1:100 dilution) was added at either 0 (low and high avidity) or 45 (medium avidity) minutes to stabilize the binding of the PE-labeled tetramer.
Staining was then analyzed using flow cytometry (LSR-II & Cytek Aurora, BD Sciences) or isolated using fluorescence-activated cell sorting (FACSAria II, BD Sciences). If cells were to be isolated, samples were immediately placed in ice and subjected to sorting at 4 °C. If cells were to be used for flow cytometry, cells were mixed into a 1-ml fix and permeabilization solution, made using the Foxp3/Transcription Factor Staining Buffer Set (Invitrogen), for 30 min on ice before intracellular staining with the indicated flow cytometry antibodies for 60 min on ice.
Operationally, T cells shedding the tetramers during the first 15 min were defined as having low avidity (Fig. 1b) and were sorted as TetPE+TetBV421−. Tetramer-associated T cells after 105 min of dissociation were defined as having high avidity and were sorted as TetPE+TetBV421+. To isolate medium-avidity T cells in a separate reaction, TetPE and TetBV421 were allowed to dissociate from T cells for 45 min to allow for the exclusion of low-avidity T cells. Anti-PE stabilizing antibody was added to the assay at t45 to prevent further dissociation of TetPE while still allowing the dissociation of TetBV421 for 30 min. Thus, medium-avidity T cells were sorted as TetPE+TetBV421− at t75 (Fig. 1b). This assay was repeated more than 100 times with reproducible results for all antigens tested.
Staining for flow cytometry and CITE-seq
Before surface staining, cells were incubated with mouse IgG isotype antibody (1:50 dilution, Invitrogen) for 20 min on ice. Cells were then stained with labeled antibodies against CD8α (clone KT15, 1:200 dilution), CD45.1 (clone A-20, 1:100 dilution), PD-1 (clone RMPI-30, 1:200 dilution), TCR-β (clone H57-597, 1:200 dilution), TIM3 (clone 5D12, 1:200 dilution), TOX (clone TXRX10, 1:50 dilution), LY108 (clone 13G3, 1:200 dilution), CX3CR1 (clone SA011F11, 1:100 dilution), pZAP70-Y292 (clone A16048B, 1:20 dilution) or TCF1 (clone C63D9, 1:50 dilution), when indicated.
Immunization and isolation of CD8+ T cells
Bone marrow isolated from the tibia and femur of syngeneic mice were cultured in complete Roswell Park Memorial Institute 1640 (RPMI 1640) medium with 20 ng ml−1 of murine granulocyte-macrophage colony-stimulating factor (PeproTech) for 1 week to generate bone marrow-derived dendritic cells (BMDCs). Mice were immunized (intradermally) with 6–9 million BMDCs pulsed with 100 µM of the indicated peptide in complete RPMI 1640 for 2 h at 37 °C. Five-to-seven days after immunization, vaccine-draining LNs were excised and isolated with the EasySep Mouse CD8+ T cell Isolation Kit followed by sorting of control (tetramer-negative), low-avidity or high-avidity cells as described in ‘Modified Tetramer Decay Assay’.
Tetramer re-decay
Cells obtained using the method described in ‘Immunization and isolation of CD8+ T cells’ were sorted as TetPE+TetBV421− and TetPE+TetBV421+ at t15 after tetramer staining and tetramer decay. We then re-stained these cells with tetramers in both colors, so that all cells were TetPE+TetBV421+ again, and subjected them to another round of tetramer decay for 15 min before analysis using flow cytometry.
Ex vivo restimulation
Ten thousand control, low-avidity or high-avidity PDPRMUT-specific T cells were isolated as described in ‘Immunization and isolation of CD8+ T cells’ and cocultured with 1,000,000 congenic splenocytes pulsed with 10 µM PDPRMUT. To determine the phosphorylation of ZAP70, cells were cocultured in complete RPMI 1640 for 5 min at 37 °C. To determine IFNγ production, cells were cocultured with 1,000,000 BMDCs pulsed with 10 µM PDPRMUT and brefeldin A (1:1,000 dilution) in complete RPMI 1640 for 4 h at 37 °C. Cells were first stained for the expression of surface molecules for 20 min on ice, then fixed and permeabilized using the Foxp3/Transcription Factor Staining Buffer for 30 min on ice before intracellular staining with the indicated flow cytometry antibodies for 60 min on ice. Staining was then detected using flow cytometry (Cytek Aurora) followed by analysis using FlowJo.
In vitro killing assay
Five thousand congenic splenocytes were pulsed with 10 µM PDPRMUT in complete RPMI 1640 for 2 h at 37 °C and used as target cells for the killing assay. Five thousand CellTrace Violet-labeled congenic splenocytes pulsed at 37 °C with dimethyl sulfoxide for 2 h and used as antigen-negative cells for the killing assay. Ten thousand control, low-avidity or high-avidity PDPRMUT-specific T cells were isolated as described in ‘Immunization and Isolation of CD8+ T cells’ and cocultured with target cells and antigen-negative cells for 4 h at 37 °C. Killing of target and bystander cells was assessed using flow cytometry (Cytek Aurora) followed by analysis using FlowJo. Specific killing, defined by the difference in the proportion of dead cells in the target population and the bystander population, is shown.
Adoptive cellular transfer
One thousand cells, isolated as in ‘Immunization and Isolation of CD8+ T cells’ were injected retro-orbitally into mice 10 days after the tumor challenge. In experiments testing the efficacy of the immune checkpoint blockade, mice receiving control, low-avidity or high-avidity cells were treated with isotype, αPD-1 or αCTLA-4, as indicated in ‘In vivo immune checkpoint blockade’, starting 13 days after the tumor challenge.
Preparation of samples for single-cell and multi-omic sequencing
Low-avidity, medium-avidity and high-avidity PDPRMUT-specific CD8+ TILs were stained with TotalSeq-C antibodies against PD-1, TIM3, LY108 and CX3CR1 (BioLegend) and sorted from 11 tumor-bearing mice as in ‘Modified tetramer decay assay’. About 6,000 cells for each condition were captured using the Next GEM Single Cell 5′ GEM Kit v2 (10X Genomics). Three libraries were generated for each condition using the Library Construction Kit, 5′ Feature Barcode Kit and Single Cell Mouse TCR Amplification Kit (10X Genomics) using the Chromium Next GEM Single Cell 5′ Reagent Kit v2 (Dual Index) (CG000330 Rev D protocol) before next-generation sequencing with the NovaSeq system (Illumina).
Preprocessing of scRNA-seq data
Sequencing data were preprocessed as described in Extended Data Fig. 5a. Briefly, separate FASTQ files for gene expression, TCR sequences and CITE-seq were generated using CellRanger55. Loom files containing RNA splicing information for each sample were then generated from FASTQ files for gene expression using Velocyto56. Outputs from these analyses were integrated using the scanpy57, scirpy58, MUON59 and scVelo60 packages in Python. Normalization of the CITE-seq antibody counts was conducted using DSB61, as implemented in the MUON package in Python. Low-quality cells (with fewer than 500 genes, greater than 5% mitochondrial gene content) were filtered out based on gene expression. The remaining cells were subjected to unbiased clustering based on their transcriptomes (Extended Data Fig. 5b). Only clusters 0, 1, 3, 4 and 8, with significantly upregulated expression of CD8+ T cell marker genes (Cd8b1, Trac and Cd3e), were used for the subsequent analysis (Extended Data Fig. 5c).
scRNA-seq processing
The remaining cells were then subjected to principal component analysis, neighbor graph analysis and UMAP graph analysis before being clustered with scanpy using the Leiden algorithm at a resolution of 0.6. The intersection of DEGs between clusters using three different statistical methods (Wilcoxon rank-sum test, t-test with overestimated variance and logistic regression) was then used to characterize the clusters. All plots for subsequent analyses were created using the scanpy and scVelo packages in Python.
Cluster annotation
The description of the clusters is based solely on DEGs (Fig. 2g) (see Supplementary Table 1 for the full list of genes). Stem-like cells expressed significantly higher memory-associated markers (Tcf7, Xcl1 and Bcl6)62,63,64,65,66 and significantly lower exhaustion-associated markers (Pdcd1, Havcr2, Lag3, Tigit, Entpd1 and Cd38)25,67,68,69,70. TEFF‐EX cells expressed significantly higher levels of transcription factors associated with effector functions (Tbx21, Ikzf2) than stem-like and TEX cells. They also expressed significantly higher levels of exhaustion-associated markers than stem-like cells, but significantly less than TEX1 cells. TEX2 cells were characterized by significantly high expression of certain markers of exhaustion (Tox, Entpd1 and Cd38), but low expression of others such as Havcr2. CD8+ T cells with a similar transcriptional profile have been reported previously9. Finally, NK-like cells were characterized by significantly high expression of NK cell receptors such as Klrc1, Klrc2, Klrd1, Klrk1 and Klre1 (Fig. 2g).
Validation of these transcriptional signatures was sought using CITE-seq with antibodies to PD-1, LY108, TIM3 and CX3CR1 (Fig. 2h). This is particularly important for genes that are poorly detected by RNA-seq (Cx3cr1 and Slamf6). Stem-like cells expressed significantly higher levels of LY108 and significantly lower levels of PD-1 and TIM3. TEFF‐EX cells expressed significantly higher levels of the effector marker CX3CR1 and significantly lower levels of TIM3. TEX1 cells expressed significantly higher levels of TIM3 and significantly lower levels of CX3CR1 and LY108. TEX2 cells expressed significantly higher levels of PD-1 and significantly lower levels of TIM3 and CX3CR1. Finally, NK-like cells expressed significantly higher levels of TIM3 and significantly lower levels of PD-1. Altogether, surface expression of these four proteins corroborated the phenotypic descriptions of the clusters based on their transcriptomes (Fig. 2g,h).
TCR sequence and trajectory inference analysis
Cells with single-chain TCRαβ were grouped into clonotypes based on identical CDR1, CDR2 and CDR3 amino acid sequences on both chains using scirpy58. Only clonotypes with more than 40 cells were used for subsequent analysis. Clonotypes were then clustered on the proportion of cells in each clonotype measured to have low, medium or high avidity using the k-mean algorithm using the scikit-learn package in Python. The resulting two clusters were deemed low-avidity and high-avidity clonotypes based on the significant difference of cells in each clonotype with low or high avidity, as measured in Fig. 1b. Trajectory inference of low-avidity and high-avidity clonotypes was then conducted using scVelo60.
Gene score development
Sequenced cells were randomly assigned to either the test or training dataset using the sampling function of pandas with frac = 0.5. Low-avidity and high-avidity T cells in the training dataset were then subjected to differential gene expression analysis with DESeq2 (avidity and cluster were used as design variables) using PyDESeq2 (ref. 42). Genes with a false discovery rate (FDR) of less than 0.1, a log2 fold change greater than 0.75 and a base mean greater than 1 were used as a gene score for the determination of low and high avidity in silico. This list was then intersected with predicted targets of TCR-related transcription factors, as annotated in the ARCHS4 database34 to develop a second gene list.
Pan-human cancer TIL atlas
Preprocessed transcriptomic data from the Pan-Cancer TIL Atlas31 was obtained from the Gene Expression Omnibus (GEO) (accession no. GSE156728). Gene scoring on all CD8+ T cells obtained was implemented using the scanpy package in Python through the scanpy.tl.score_genes function with the genes described in ‘Gene score development’ as inputs for the gene_list parameter. Cells one s.d. below and above the mean of the gene score were deemed low and high avidity, respectively. Differential gene expression analysis using DESeq, with avidity as the only design variable, was used to compare low-avidity and high-avidity T cells. DEGs (as defined in ‘Gene score development’) were plotted using the volcano function of PyDESeq2 (ref. 42).
Immune checkpoint blockade response prediction
Transcriptomic data of pretreated TILs was obtained from the GEO and European Genome-phenome Archive (EGA) database for patients with hepatocellular carcinoma35 (accession no. EGAS00001007547), melanoma36(accession no. GSE120575) and triple-negative breast cancer37 (accession no. GSE169246). CD8+ T cell clusters, as defined using the Leiden algorithm, were then extracted to determine avidity in silico using the methods described in ‘Pan-human cancer TIL atlas’. The proportion of low-avidity and high-avidity CD8+ T cells was then compared between responders and nonresponders using a two-way ANOVA.
Statistical methods
Statistical tests were conducted using Prism (GraphPad Software). Statistical tests included paired t-tests, unpaired t-tests, one-way ANOVA, two-way ANOVA and Mantel–Cox survival analysis. Paired t-tests were used in place of unpaired t-tests when comparing matched low-avidity and high-avidity T cells derived from the same samples. When present, error bars denote the s.d. of samples.
No statistical methods were used to predetermine sample sizes, but our sample sizes are similar to those reported in previous publications17. Individual data points are shown in each figure; data distribution was assumed to be normal but this was not formally tested. For the purpose of randomization, all mice that participated in an experiment were placed in a single container; they were randomly picked and assigned sequentially to individual groups. No data were excluded from the analyses. Blinding was not necessary because no data were excluded and only objective measurable criteria were used.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.

留言 (0)