The present study was conducted in compliance with all relevant ethical regulations, with approval from the University of North Carolina at Chapel Hill (UNC) Animal Husbandry and Institutional Animal Care and Use Committee (IACUC). Data collection and analysis were not performed blind to the conditions of the experiments. Data points were excluded from the analysis in experiments in which positive control failed.
Mouse
Male and female 6- to 8-week-old NSG (NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ) mice were purchased from the Animal Core Facility at the UNC. Mice were housed in the Animal Core Facility at the UNC with specific pathogen-free conditions of a 12 h dark:light cycle, 23 °C ambient temperature and 50% humidity. All mouse experiments were performed in accordance with UNC Animal Husbandry and IACUC guidelines and were approved by the UNC IACUC.
Primary human T cells
Buffy coats from healthy donors were purchased from the Gulf Coast Regional Blood Center under an institutional review board (IRB)-exempt protocol. Buffy coats were deidentified and donor information (sex, age and so on) were not disclosed. PMBCs were isolated by Ficoll centrifugation (Axis-Shield). Samples deidentified and obtained from patients with CLL were acquired from the UNC Tissue Procurement Facility (IRB no. 09-0768).
Cell lines and culture
HEK293T, Daudi and Jurkat cells were purchased from American Type Culture Collection (ATCC). The neuroblastoma cell lines LAN-1 and CHLA-255 were gifts from M. Brenner and L. Metelitsa, respectively, of Baylor College of Medicine. Tumor cell lines were transduced with a γ-retrovirus vector encoding FFLuc. All tumor cell lines were maintained in complete Roswell Park Memorial Institute-1640 medium (Gibco) supplemented with 10% fetal bovine serum (FBS, Thermo Fisher Scientific), 2 mM GlutaMAX (Gibco), 100 U ml−1 of penicillin and 100 μg m−1 of streptomycin (Gibco). Cell lines were routinely tested for Mycoplasma and cell-surface expression of target antigens and kept in culture for <2 months consecutively, after which a new aliquot was thawed. Cell lines used in the present study were not on the International Cell Line Authentication Committee crosscontamination list.
Automated high-throughput screening of KIs in activated T cells
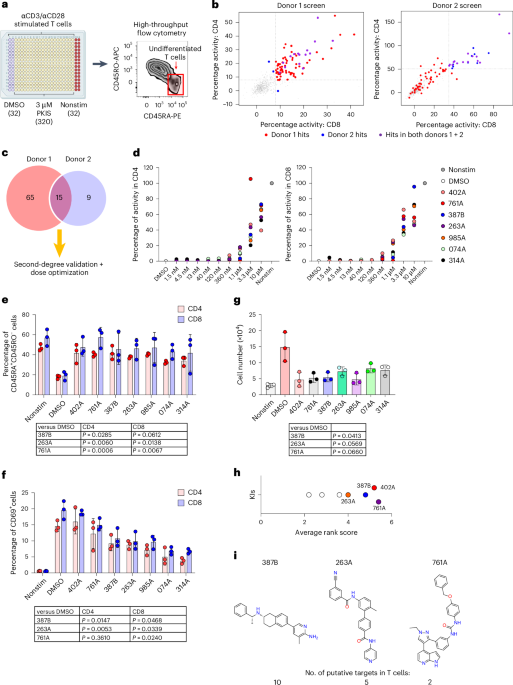
We screened KIs contained in a kinase-focused set of compounds (PKIS1 and PKIS2, including 810 compounds, which are a subset of the PKIS collection) using a 384-well plate pre-stamped for 3 μM final screening concentration (https://www.sgc-unc.org/kinase-chemogenomics) that has been previously successfully used for high-throughput screening despite each KI having a unique half-maximal inhibitory concentration (IC50)50,51. This plate was used to seed T cells isolated from the peripheral blood of healthy donors (Gulf Coast Regional Bank) in the presence of the Immunocult human CD3 or CD28 T cell activator (25 μl per 106 ml of cells) for 5 d without any addition of exogenous cytokines. T cell differentiation was evaluated by measuring the phenotypic transition from CD45RA+ to CD45RO+ in both CD8 and CD4 T cells. Nonstimulated T cells and T cells stimulated in the presence of DMSO served as controls. After staining with CD8, CD45RA and CD45RO antibodies, flow cytometry data were acquired on BD LSRII with high-throughput screening mode. Data were then uploaded and analyzed in the CICBDD searchable database system (ScreenAble Solutions; https://www.screenablesolutions.com). To enable hit compound selection, we applied two exclusion criteria to filter flow cytometry data and remove hits causing T cell death and hits that blocked T cell activation or proliferation. We first calculated the fold expansion of the cell number in each test well at day 5, based on the cell number of an inner-plate, nonstimulated control and test compounds that led to excessive cell death or impaired proliferation (fold expansion <0.75) were removed from the analysis. Second, we used mean intensity of FSC on live events (cell blasting) as a secondary readout for T cell activation and test compounds that reduced FSC by >30% (compared with DMSO control) at day 5 of culture were also excluded. To normalize for the variations between blood donors and experiments, we presented the percentage of activity for each compound based on the percentage CD45RA+CD45RO− cells for each compound (readoutcompound) and for inner-plate controls (readoutDMSO and readoutnonstim) with the following formula: Percentage activity = ((readoutcompound − readoutDMSO)/(readoutnonstim − readoutDMSO)) × 100%.
To pick the final three KIs for downstream analysis, we score ranked each compound based on their ability to block T cell differentiation (based on percentage CD45RA+CD45RO− in CD4 or CD8 T cells), impact on T cell activation (based on CD69 staining in CD4 or CD8 T cells) and T cell numerical counts. Each phenotype was scored 1–7. with the top-ranked KI scoring 7. We then averaged the ranked score (five scores) for each compound to determine the best candidates, as previously described29.
MIB enrichment coupled to MS analysis
Jurkat cells were treated with DMSO or KIs at the concentration of 0.75 μM or 2.0 µM for 1 h at 37 °C. Cells were then lysed on ice in MIB lysis buffer (50 mM Hepes, pH 7.5, 0.5% Triton X-100, 150 nmol of NaCl, 1 mM (ethylenebis(oxonitrilo))tetra-acetate, 10 mM sodium fluoride, 2.5 mM sodium orthovanadate and 1× protease inhibitor cocktail) (Roche) and 1% phosphatase inhibitor cocktail (Sigma-Aldrich) and treated as previously described39. Briefly, cell lysates were sonicated on ice and then centrifuged to obtain supernatant preparations. The filtered lysate was passed through a column of layered inhibitor-conjugated beads (MIBs). The columns were washed and kinases eluted with sodium dodecylsulfate. Eluted kinases were resuspended in 50 mM Hepes, pH 8.0, reduced and alkylated and digested with sequencing grade-modified trypsin (Promega). Digested peptides were analyzed by liquid chromatography–tandem MS38,52. Raw data were processed using the MaxQuant software suite (v.1.6.15.0) for identification and label-free quantification (LFQ)53. Data were searched against a Uniprot Human database (containing 20,350 sequences) using the integrated Andromeda search engine. A maximum of two missed tryptic cleavages were allowed. The fixed modification was set to: cysteine carbamidomethylation; the variable modifications were set to: methionine oxidation and amino-terminal acetylation. LFQ and match between runs were enabled. Results were filtered to 1% false recovery rate (FDR) at the unique peptide level. Kinases were parsed and global normalization was applied. Perseus was used for further processing54. Only kinases with more than one unique + razor peptide were used for LFQ analysis. To identify potential kinase targets for each inhibitor, we first removed all metabolic kinases from our analysis because they often show atypical responses in the assay. Then, log2(fold-change ratios) (compared with DMSO) were calculated using the averaged log2(LFQ intensities) and kinases with log2(fold-change) ≥ 0.5 were considered potential targets for further validation. We also excluded a few kinases that exhibit a reverse dose effect (the 0.75 μM treatment condition showed more profound effect on kinase enrichment than the 2.0 μM condition).
Plasmid construction, virus production and generation of CAR T cells
The retroviral vectors encoding the CAR.CD19 and CAR.GD2 with the CD28 or 4-1BB endodomains and the methodology to generate retroviral supernatants have been previously reported19. The lentivirus constructs encoding shRNAs targeting CLK3, STK17B, MAP3K7, AURKA, MINK1, CDK12, TAOK2, ITK, CDK13, ADCK3, ADCK1, MAP3K4 and TRIM28 were obtained from the UNC shRNA Core Lab. Lentiviral supernatant was generated by transient transfection of 293T cells with lentiviral vector, ps.pAX2 plasmid encoding gag-pol and pMD.2G plasmid encoding the vesicular stomatitis virus G envelop. Lentivirus supernatant was concentrated using the concentrator (Takara Bio) according to manufacturer’s instructions. Mononuclear cells were separated by lymphoprep density separation, activated in plates coated with 1 μg ml−1 of anti-CD3 (Miltenyi Biotec) and 1 μg ml−1 of anti-CD28 (BD Biosciences) mAb, transduced with either retroviral vectors or lentiviral vectors and expanded in medium with recombinant IL-7 (10 ng ml−1) and IL-15 (5 ng ml−1) (PeproTech)3,55. KIs were added to the culture and DMSO at an equal volume was used as a control. At day 10 post-transduction, T cells were collected, washed, rested from cytokines for 24 h and then used for in vitro and in vivo mouse tumor model experiments. To generate CAR T cells co-expressing specific shRNA, activated T cells were co-transduced with retroviral supernatant encoding the CAR and lentiviral supernatant encoding the shRNA. At day 3 post-transduction, CAR T cells were selected using 1.0 μg ml−1 of puromycin for 48 h. At day 5, CAR T cells were washed and further expanded in complete medium with IL-7 (10 ng ml−1) and IL-15 (5 ng ml−1) and tested for functionality at day 10 (ref. 36). For the experiments in which naive cells and memory cells were used as starting cell subsets, we isolated them from mononuclear cells according to the manufacture’s protocol (Miltenyi Biotec) and used them to generate CAR T cells as described for CAR T cells generated from total PBMCs.
In vitro co-culture assays
Co-culture experiments with tumor cells and CAR T cells were performed as previously described19. CAR T cells were extensively washed and maintained in complete medium without DMSO or KIs for 24 h before testing them in co-culture experiments. Supernatants were collected after 24 h of co-cultures to measure specific cytokines by ELISA. In selected experiments, CAR T cells were stimulated with phorbol 12-myristate 13-acetate, brefeldin A and ionomycin (Thermo Fisher Scientific) and specific cytokines detected by intracellular staining according to the manufacturer’s instructions (BD Biosciences). For the proliferation assay, CAR T cells were labeled with 5 μM CellTrace Violet (CTV, Invitrogen) according to the manufacturer’s instruction, co-cultured with tumor cells for 5 d, then collected and analyzed by flow cytometry.
Flow cytometry
CAR.CD19 and CAR.GD2 expression on T cells was detected using anti-idiotype antibody followed by Alexa Fluor 647 (AF647)-conjugated, secondary goat, anti-mouse immunoglobulin G (IgG; clone A8 5-1, BD Bioscience)37,56. For the detection of TCF1, CAR T cells were fixed and permed after cell-surface staining and then stained with the PE- or BV421-conjugated TCF1 antibody (clone S33-966, BD Biosciences) diluted in True-Nuclear 1× Perm Buffer for 30 min at 25 °C according to manufacturer’s instruction (BioLegend). The following antibodies were used for the phenotypic characterization of T cells and tumor cells: BV421-conjugated anti-CD4 (clone RPA-T4, BD Biosciences), BV711-conjugated anti-CD4 (clone SK3, BD Biosciences), Alexa Fluor-700-conjugated anti-CD8 (clone RPA-T8, BD Biosciences), allophycocyanin (APC)-conjugated anti-CD45RO (clone UCHL1, BD Biosciences), PE-conjugated anti-CD45RA (clone HI100, BD Biosciences), V500-conjugated anti-CD45RA (clone HI100, BD Biosciences), APC-H7-conjugated anti-CD3 (clone SK7, BD Biosciences), PE-conjugated-anti-TNF (clone MAb11, BD Biosciences), BV786-conjugated anti-IFNγ (clone 4S.B3, BD Biosciences), FITC-conjugated IL-2 (clone 5344.111, BD Biosciences), FITC-conjugated anti-CCR7 (clone 150503, BD Biosciences), FITC-conjugated anti-CD20 (clone L27, BD Biosciences), APC-conjugated CD19 (clone HIB19, BD Biosciences), PE-conjugated GD2 (clone 14.G2a, BD Biosciences), BV421-conjugated anti-CD69 (clone FN50, BD Biosciences), FITC-conjugated CD69 (clone L78, BD Biosciences), PE-conjugated CD95 (clone DX2, BD Biosciences), BV605-conjugated CD62L (clone DREG-56, BD Biosciences), PE-conjugated CD122 (clone Mik-β2, BD Biosciences), PE-conjugated CD127 (clone HIL-7R-M21, BD Biosciences), PE-conjugated CD137 (clone 4B4-1, BD Biosciences), PE-CY7-conjugated CD25 (clone 2A3, BD Biosciences), BV421-conjugated CD27 (clone M-T271, BD Biosciences), PE-CY7-conjugated CD28 (clone CD28.2, BD Biosciences), PE-CY7-conjugated CD279 (clone EH12.1, BD Biosciences), BV711-conjugated TIM3 (clone 7D3, BD Biosciences), PE-conjugated LAG3 (clone T47-530, BD Biosciences), AF647-conjugated TOX (clone NAN448B, BD Biosciences), PE-CY7-conjugated ZAP70-pY319 (clone 17A/P-ZAP70, BD Biosciences), BV605-conjugated mouse IgG1, κ isotype control (clone X40, BD Biosciences), PE-conjugated mouse IgG1, κ isotype control (clone MOPC-21, BD Biosciences), PE-conjugated mouse IgG2b and κ isotype control (clone MPC-11, BioLegend). All antibodies were used at a 1:100 dilution (in phosphate-buffered saline (PBS)) for staining. Flow cytometry data were acquired by SD LSRFortessa (BD Biosciences) and analyzed by using FlowJo software (FlowJo v.10, LLC).
Immunoblot analysis
Whole-cell protein lysates were obtained from cells treated with radioimmunoprecipitation assay lysis and extraction buffer (Thermo Fisher Scientific) supplemented with proteinase and phosphatase inhibitors (Thermo Fisher Scientific). Equivalent amounts of proteins were mixed with 4× loading buffer (BioRad) and separated on precast 4–15% gradient polyacrylamide gel electrophoresis gels (BioRad). Proteins were then transferred to poly(vinylidene fluoride) membranes (BioRad) and signals were detected by SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Fisher Scientific) or SuperSignal West Pico Plus Chemiluminescent Substrate (Thermo Fisher Scientific). Actin or CD3ζ was used as a loading control. The following antibodies were used: anti-CD3ζ (Santa Cruz, cat. no. sc166275), anti-actin (Santa Cruz, cat. no. sc47778), anti-MINK1 (Thermo Fisher Scientific, cat. no. PA5-51000), anti-AURKA (Cell Signaling Technology, cat. no. 14475s), anti-CLK3 (Cell Signaling Technology, cat. no. 3256), anti-STK17B (Proteintech, cat. no. 26600-1-AP), anti-MAP3K7 (Cell Signaling Technology, cat. no. 4505), anti-CDK12 (Cell Signaling Technology, cat. no. 11973), anti-TAOK2 (Thermo Fisher Scientific, cat. no. 21188-1-AP), anti-CDK13 (Invitrogen, cat. no. PA5-27702), anti-ADCK3 (Abcam, cat. no. ab230897), anti-ADCK1 (Sigma-Aldrich, cat. no. HPA051012), anti-KAP1 (TRIM28) (Abcam, cat. no. ab10484), anti-ZAP70 (Cell Signaling Technology, cat. no. 3165S), anti-phospho-Zap70 (Tyr319)/Syk (Tyr352) (Cell Signaling Technology, cat. no. 2701L), anti-LAT (Cell Signaling Technology, cat. no. 45533S), anti-phospho-LAT (Tyr171) (Cell Signaling Technology, cat. no. 3581), anti-p38 (Cell Signaling Technology, cat. no. 8690), anti-phospho-p38 Thr180/Tyr182 (Cell Signaling Technology, cat. no. 4511), anti-FOXO1 (Cell Signaling Technology, cat. no. 2880), goat anti-rabbit IgG-horseradish peroxidase (HRP) (Invitrogen, cat. no. 656120), rabbit anti-mouse IgG-HRP (Invitrogen, cat. no. 31430). All antibodies were used at 1:1,000 dilution (in Tris-buffered saline with Tween 20 containing 3% skimmed milk). Immunoblot images were acquired with Chemi Doc MP system (BioRad).
In vivo mouse tumor models
NSG mice were injected intravenously with Daudi or CHLA-255 tumor cells labeled with the FFLuc gene37,56. Then 5–14 d after tumor engraftment, mice were randomly assigned into control or experiment groups and infused i.v. with natural T (NT) or CAR T cells manufactured in the presence of either DMSO or KIs. In selected experiments, mice were euthanized after 12 d or 17 d after CAR T treatment to measure T cell expansion in vivo by flow cytometry. In the experiments designed to evaluate the antitumor effects, tumor bioluminescence (BLI) was measured over time using IVES Kinetic (PerkinElmer), IVIS spectrum (PerkinElmer) or AMI HT (Spectral) in vivo imaging systems. Mice were euthanized when developing signs of discomfort as per protocol and as recommended by veterinarian and maximal tumor size or burden was not exceeded. In selected experiments tumor-free mice after treatment with CAR T cells were rechallenged with the same number of tumor cells.
Assay for ATAC–seq
ATAC–seq profiling performed by Active Motif used 105 cells. Briefly, purified TN cells and CAR T cells generated from TN cells from two healthy donors, and then exposed to either DMSO or KIs for 72 h and 105 cells, were analyzed. We achieved between 7.9 × 107 and 9.3 × 107 total reads per sample, and the total number of peaks ranged from 11.9 × 103 to 22.4 × 103 per sample. For comparative analysis, we downsampled the number of tags in each sample to the sample with the fewest usable number of tags (1.1 × 107). Peaks were then called using MACS with a cutoff P value of 1 × 10−7 (-nomodel option)57. For peak filtering, we removed false peaks within the ENCODE blacklist and the fraction of peaks in reads were all >10%, except for the naive sample for patient 2, which was 4.53%. PCA plots were generated based on all variable regions and representative browser tracks were generated in Integrative Genomics Viewer.
RNA-seq analysis
Total RNA was isolated from roughly 2 × 106 CAR.CD19 T cells or FACS-sorted TE cell/TEM cell (CD45RA+/−CCR7−) and TSCM cell (CD45RA+CCR7+) subsets using QIAGEN’s RNeasy Plus Mini Kit (cat. no. 74134) according to the manufacturer’s instructions. RNA quality was assessed using the Agilent 2200 TapeStation system. RNA-seq was performed using Illumina’s Stranded mRNA Prep Ligation Kit (cat. no. 20040534) for library preparation, starting with 100 ng of total RNA. This method uses oligo(dT) magnetic beads to capture mRNAs with poly(A) tails. The average fragment length for these libraries was between 300 bp and 400 bp. Paired-end sequencing was performed using Illumina’s NextSeq 2000 kit v.3.0 (2 × 100), obtaining at least 27 million reads per sample. Sequencing reads were aligned to the human genome (hg38) with the STAR aligner (v.2.7.6a) and subsequently quantified with Salmon (v.1.4.0). Differential expression analysis was conducted with DESeq2 (v.1.38.3) running on R (v.4.2.2). For comparison between FACS-sorted TE or TEM cells and TSCM cells in CAR.CD19 T cells, genes with log2(fold-change) > 0.8 or < −0.8 and an FDR < 0.05 were considered to be significant. Genes that significantly upregulated within TSCM cells were analyzed for upstream regulators (TFs) using ChEA3 (ref. 40). For comparison between DMSO- and KI-treated CAR.CD19 T cells or FACS-sorted TSCM cells, genes with log2(fold-change) > 0.7 or < −0.7 and an FDR < 0.05 in comparisons of DMSO and KI groups were considered to be significant. GSEA was performed with GSEA v.2 software (Broad Institute) on genes that were differentially expressed between KI and DMSO groups at different time points, respectively, against gene sets specified in each figure. Over-representation analyses were performed at https://www.gsea-msigdb.org/gsea/msigdb/human/annotate.jsp using genes that are up- or downregulated at both 24 h and 72 h in KIs versus the DMSO group. Figures were plotted using ggplot2 (v.3.40) and ComplexHeatmap (v.3.18) R package.
Quantitative real-time (RT-)PCR
Total RNA was extracted using an RNeasy plus mini kit (QIAGEN) and reverse transcription was carried out by SuperScript VILO Master Mix (Invitrogen) according to the manufacturer’s protocol. Quantitative (q)PCR was performed in triplicate with SYBR Green Master Mix (Thermo Fisher Scientific) on a QuantStudio 6 Flex Real-Time PCR system (Applied Biosystem) per the manufacturer’s instructions. The primers were used to detect the indicted gene expression and 18S was set as the control (GENEWIZ).
Statistical analysis
Student’s t-test or two-way analysis of variance (ANOVA) was used to compare groups. A nonparametric test was used for comparisons when normality assumption could not be verified. All statistical analyses were performed as two-tailed tests. We reported raw P values. CAR T cells for the in vitro experiments were generated from three or more different heathy donors. Each in vitro experiment was performed at least 3×. In vivo mouse experiments were performed with CAR T cells generated from two or more donors and performed twice independently to ensure reproducibility and completeness. Each dot represents one donor in in vitro experiments and one mouse in in vivo mouse experiments. Graph generation and statistical analyses were performed using GraphPad Prism v.8 (GraphPad Software). No statistical methods were used to predetermine sample sizes, but our sample sizes are similar to those reported in previous publications36,58. P < 0.05 was considered to be statistically significant.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.

留言 (0)