
記住我
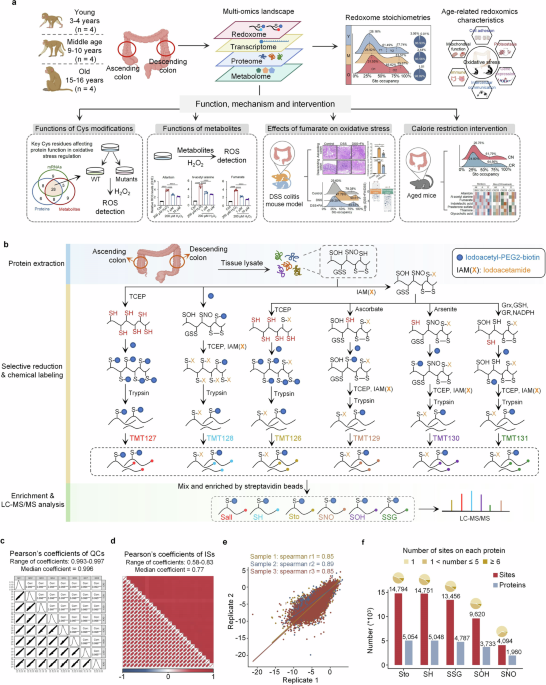

Macaca fascicularis were housed in a uniformly controlled laboratory in Canton and maintained at approximately 25 °C with a 12-h light and 12-h dark cycle. All the animals had no previous clinical or experimental background that could affect their natural aging process or increase their vulnerability to illnesses. Tissues from the colons of 4 young (females, 3–4 years old), 4 middle-aged (females, 9–10 years old), and 4 old (females, 15–16 years old) Macaca fascicularis were excised and immediately frozen in liquid nitrogen for preservation. This study adhered to the ethical guidelines for the treatment of non-human primates and was approved by the Institutional Animal Care and Use Committee at Yuanxi Biotech Inc. in Guangzhou (approval number: YXSW-2016-01).
C57BL/6J mice were purchased from GemPharmatech Co., Ltd. (Jiangsu, China). All the animals were kept in a specific pathogen-free (SPF) animal facility with a 12 h photoperiod. Before the experiment, the mice were cohoused, provided with a normal chow diet, and housed with ad libitum access to water. For the calorie restriction (CR) experiments, the mice were randomly grouped. The utilization and welfare of the mice were authorized by the Animal Experiment Ethics Committee of the State Key Laboratory of Biotherapy at Sichuan University (approval number: 20230307027).
Procedures for redoxomicsFrozen colon tissues from monkeys or mice were lysed with HEPES buffer (10 mM EDTA, 250 mM HEPES, 0.1 mM neocuproine, pH 7.5, 0.5% SDS, and 1% protease inhibitor cocktail) and homogenized twice via gentleMACSTM following the procedure of protein_01.01. The crude lysate was then sonicated in an ice water bath for 5 min, followed by centrifugation at 20,000×g for 30 min at 4 °C. The supernatant was obtained, and the concentration was determined via a bicinchoninic acid (BCA) assay (23227, Thermo Fisher Scientific).
To label the free Cys residues (SH) with a biotin reagent, the protein was diluted to 1 μg/μL in reaction buffer (5 mM EDTA, 50 mM Tris HCl, pH 8.3). Then, the mixture was incubated with 0.8 mM iodoacetyl-PEG2-biotin (21334, Thermo Fisher Scientific) in the dark for 90 min at room temperature. Afterward, the mixture was precipitated with methanol/water/chloroform. Following precipitation, 50 μL of resuspension buffer (250 mM HEPES, pH 7.7, 0.1% SDS, and 8 M urea) and 50 μL of reaction buffer were added. The protein concentration was quantified again via the BCA assay. Next, 5 mM Tris(2-carboxyethyl) phosphine (TCEP, C4706, Sigma-Aldrich) was added, and the mixture was incubated at 56 °C for 1 h. Subsequently, 10 mM iodoacetamide (IAM, I1149, Sigma-Aldrich) was added, and the mixture was incubated in the dark for 30 min at room temperature to allow for alkylation. After the reaction, 50 μg of protein was isolated, precipitated with methanol/water/chloroform, and digested with sequencing-grade modified trypsin (V5117, Promega).
To label Cys total oxidation (Sto) with a biotin reagent, the protein sample was mixed with 10 mM IAM and incubated to block free thiol groups. The protein was then precipitated to remove excess IAM. Next, the protein was redissolved in 50 μL of resuspension buffer and 50 μL of reaction buffer, followed by reduction with 5 mM TCEP. To remove excess TCEP, the protein was precipitated again and subsequently redissolved in 50 μL of resuspension buffer and 50 μL of reaction buffer. The concentration was reassessed via the BCA assay. Iodoacetyl-PEG2-biotin (1.2 mM) was added, and the mixture was incubated for 90 min. Finally, 50 μg of protein was precipitated and then digested with trypsin for 16 h.
To label Cys residues with SOH or SNO modifications with a biotin reagent, the protein was diluted to 1 μg/μL with 100 mM TEAB. A total of 100 μg of protein was subsequently combined with 10 mM IAM to block free thiol groups. The protein was precipitated via methanol/water/chloroform and then resuspended in 50 μL of HENS buffer (1 mM EDTA, 250 mM HEPES, pH 7.7, 0.1 mM neocuproine, and 1% SDS) and 50 μL of reaction buffer. Next, 20 mM sodium arsenite (for SOH) or sodium ascorbate (for SNO) and 0.4 mM iodoacetyl-PEG2-biotin were added and incubated. To remove excess reagents, precipitation was performed. The protein was redissolved in 50 μL of resuspension buffer and 50 μL of reaction buffer, and its concentration was reassessed using the BCA assay. The protein was subsequently reduced by incubation with 5 mM TCEP, followed by alkylation with 10 mM IAM. Finally, 50 μg of protein was precipitated, and digestion was carried out with trypsin.
To label Cys residues with SSG modification with a biotin reagent, 100 μg of protein lysates were diluted to 1 μg/μL with 100 mM TEAB and treated with 10 mM IAM. After blocking, the protein was precipitated with methanol/water/chloroform and then redissolved in a resuspension buffer. Buffer exchange was performed using 0.5 mL Amicon Ultra 10 K filter units (Millipore, Burlington, MA, USA) with exchange buffer (25 mM HEPES, 1 M urea, pH 7.5). The protein concentration was quantified via the BCA method. The samples were diluted to a concentration of 1 μg/μL with exchange buffer. To reduce the SSG, the following components were added to the solution: 2.5 μg/μL glutaredoxin 1 M (Grx1M, C14S), 0.25 mM oxidized glutathione (GSSG, G8690, Solarbio), 4 U/mL glutathione reductase (GR, P2372S, Beyotime), and 1 mM reduced nicotinamide adenine dinucleotide phosphate (NADPH, ST360, Beyotime). The samples were maintained at 37 °C for 10 min to allow for incubation, quickly chilled on ice, and transferred to a 0.5 mL Amicon Ultra 10 K filter. Excess reagents were removed by washing with reaction buffer to a final volume of 100 μL, and the concentration was quantified using a BCA method. The sample was then reacted with 0.4 mM iodoacetyl-PEG2-biotin and precipitated with methanol/water/chloroform. After precipitation, 50 μL of resuspension buffer and 50 μL of reaction buffer were added. The protein concentration was reassessed via the BCA assay. The sample was then reduced with 5 mM TCEP and alkylated with 10 mM IAM. Finally, 50 μg of protein was precipitated and digested with trypsin.
To label all the Cys residues with a biotin reagent, equal protein amounts from each sample were taken and combined to create a mixed sample. The concentration was assessed via the BCA method. The mixed sample was diluted to 1 μg/μL with 100 mM TEAB, reduced with 5 mM TCEP, and precipitated to remove excess TCEP. The pellet was dissolved in 50 μL of resuspension buffer and 50 μL of reaction buffer, and the protein concentration was again assessed via BCA. Next, alkylation was carried out with 2 mM iodoacetyl-PEG2-biotin. The protein was then precipitated with methanol/water/chloroform and subjected to digestion with trypsin.
TMT labeling for redoxomicsRedoxomic analysis of Macaca fascicularis and mouse colon tissue samples was performed via a tandem mass tag (TMT)-based strategy. Each batch of TMT-labeled samples included a shared reference sample, which was biotin labeled with Cys residues (Sall) and labeled with TMT-127. The other five channels of the TMT-6plex reagent (Thermo Scientific) were assigned to label five different Cys redox states of the same sample: TMT-126 for Sto, TMT-128 for SH, TMT-129 for SNO, TMT-130 for SOH, and TMT-131 for SSG. For each TMT channel, 10 μg of peptide was isobarically labeled. The six channels, with a total of 60 μg of peptide, were combined and then dried via a SpeedVac. The dried peptides were desalted via a C18 solid-phase extraction (SPE) column (10 mg, Waters). Notably, in the redoxomic analysis of colon tissues from DSS-induced mice, the reference sample (Sall) was labeled with TMT-127. The free SH groups in the ascending colon samples were labeled with TMT-126, the Sto groups in the ascending colon samples were labeled with TMT-129, the free SH groups in the descending colon samples were labeled with TMT-130, and the Sto groups in the descending colon samples were labeled with TMT-131.
Enrichment of biotinylated peptides using streptavidin magnetic beadsLabeled peptides (60 μg) were dissolved in 60 μL of 50 mM TEAB. Among these, 4 μg was desalted and subjected to LC-MS/MS analysis. This measurement was intended to determine the protein amounts in each channel, which would be utilized for normalizing the redox modification ratios. The remaining biotin-labeled peptides were enriched via streptavidin magnetic beads (HY-K0208, MCE). The mixture was then incubated at 4 °C for 6 h. The streptavidin magnetic beads were washed sequentially once with 1 mL of PBST (150 mM sodium chloride, 20 mM potassium phosphate, 0.5% Tween-20, pH 7.2), 500 μL of 1 M NaCl, 2 mL of PBST, 500 μL of PBS, and 500 μL of ddH2O. Finally, 100 μL of 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP, 105228, Sigma-Aldrich) was used to elute the biotinylated peptides at 59 °C for 5 min, and this elution step was repeated 3 times. All eluates were combined and concentrated to dryness. The eluted samples were then desalted via a C18 ZipTip and analyzed via LC-MS/MS.
Sample preparation and TMT labeling for proteomicsProtein extraction, reduction, alkylation, and enzymatic digestion of colon tissues were carried out as previously described.32 A proteomic analysis of mouse colon tissues employed a TMT-based approach, where every batch of TMT-labeled samples included a shared reference sample. For calorie-restricted mice, the reference sample was tagged with TMT-127N. Twenty samples were tagged using the other ten channels of the TMT-11plex reagents (Thermo Scientific). The labeling process adhered to the instructions outlined in the TMT kit, with each sample containing 70 μg of peptides being labeled. After quenching with 5% hydroxylamine, the samples were combined and used for proteomics analysis. For the fumarate treatment of DSS-treated mice, the reference sample was tagged with TMT-128C. Eighteen samples were tagged via the other nine channels of the TMT-10plex reagents (Thermo Scientific). The labeling procedure followed the TMT kit instructions and used 10 μg of peptides per sample. The labeled peptides were redissolved in 0.1% TFA and desalted with C18 SPE columns (10 mg, Waters), followed by fractionation with high-performance liquid chromatography (RP-HPLC; LC2030 plus; Shimadzu). Briefly, a total of 50 μg of TMT-labeled peptides were fractionated via a Poroshell HPH C18 column (250 × 4.6 mm, OD 4 μm, Agilent) at a flow rate of 1 mL/min. The fractionation process involved the use of solvent A (98% water, 2% acetonitrile, pH 10) and a gradient increase in solvent B (10% water, 90% acetonitrile, pH 10). The 120-min LC gradient was set as follows: 0–2% B in 2 min, 2–35% B in 58 min, 35–55% B in 35 min, 55–90% B in 21 min, and 90–2% B in 4 min. The initially obtained 120 fractions were combined into 20 fractions, which were subsequently dried and desalted via Zip-tip columns.
Procedures for untargeted metabolomicsApproximately 30 mg of mouse colon tissue and four steel beads were placed in 500 μL of precooled 80% methanol. The tissue was homogenized via Precellys Evolution Touch at 6800 rpm for 10 s, followed by a 30 s pause, and this process was repeated three times. Afterward, 500 μL of 80% methanol was introduced into the mixture and vortexed at 1500 rpm for 30 s. The lysate was then incubated at −80 °C for 30 min, ultrasonicated in an ice water bath for 10 min, and centrifuged at 13,000 rpm for 20 min at 4 °C. Following centrifugation, 600 μL of the supernatant was obtained, and 500 μL of 80% methanol was added again. The mixture was vortexed and centrifuged, and the supernatants were combined. Additionally, 200 μL of extracted metabolites from each sample were pooled to serve as the QC for mass spectrometry (MS) analysis. The supernatants from all the samples were concentrated and evaporated under a vacuum. The protein precipitate was dried and redissolved in 0.1 M KOH, and the concentration was determined via BCA for subsequent data calibration.
LC-MS/MS analysisFor the analysis of the enriched biotinylated peptides, an EASY-nanoLC 1000 system (Thermo Fisher Scientific) coupled with a Q Exactive Plus mass spectrometer (also from Thermo Fisher Scientific) was used to perform LC-MS/MS analysis. The desalted peptides were reconstituted in buffer A (98% water, 2% acetonitrile, 0.1% formic acid) and separated via a custom trap column (2.5 cm × 75 μm) filled with Spursil C18 particles (5 μm; DIKMA), followed by an analytical column (25 cm × 75 μm) filled with Reprosil-Pur C18-AQ particles (1.9 μm; Dr Maisch). The separation was accomplished with buffer A and a nonlinear increase in buffer B (80% acetonitrile, 0.1% formic acid) using a gradient ranging from 14% to 100% across 120 min with a flow rate of 330 nL/min. Data-dependent acquisition (DDA) was carried out using Xcalibur software. The MS1 full scan was set to a range from 350 m/z to 1600 m/z, with a resolution of 70,000, the automatic gain control (AGC) set at 3e6, and the maximum ion injection time (IT) at 20 ms. Fragmentation was performed on the 15 most abundant parent ions using a normalized collision energy (NCE) of 30%. For MS2 analysis, a resolution of 35,000 was applied, with an AGC target of 1e5 and a maximum IT of 100 ms. An isolation window of 0.6 m/z was applied, and precursors with unassigned charge states or charge states of z = 1, 6–8, or unassigned charge states were not considered for further analysis.
To analyze the unenriched peptides for protein quantification in each channel, LC-MS/MS analysis was performed via the same instrumentation setup. A 65-min gradient, ranging from 13% to 100% Buffer B, was used for the separation of peptides at a flow rate of 330 nL/min. DDA was conducted in positive ion mode, and the parameter settings for MS1 and MS2 were the same as those described above.
For proteomics, LC-MS/MS analysis was carried out via an EASY-nanoLC 1000 (Thermo Fisher Scientific) coupled to a Q Exactive Plus mass spectrometer (Thermo Fisher Scientific). The desalted peptides were reconstituted in Buffer A. The peptides were separated via a custom trap column (2.5 cm × 75 μm) filled with Spursil C18 particles (5 μm; DIKMA) and an analytical column (25 cm × 75 μm) filled with Reprosil-Pur C18-AQ particles (1.9 μm; Dr. Maisch). Peptides were eluted with a 90-min gradient from 13% to 100% Buffer B with a flow rate of 330 nL/min. DDA was conducted using Xcalibur software. The parameter settings for MS1 and MS2 were identical to those described above.
Hydrophilic metabolites extracted from mouse colon tissues were analyzed via an Ultimate 3000 UHPLC (Dionex) coupled with a Q Exactive mass spectrometer (Thermo, CA). The temperature of the column was maintained at 35 °C. The specific parameters for metabolite detection were consistent with those used in previous methods.32
MS database searchingSpectrometric raw data files from redoxomics and proteomics were processed using MaxQuant (version 1.6.1.0). The omics data from Macaca fascicularis colon tissues were aligned against a combined Macaca fascicularis protein sequence database, which contained 76,242 protein sequences. For mice, the omics data were aligned to the Swiss-Prot mouse protein sequence database, updated on 07/2019, which consists of 17,019 sequences. TMT-6plex was employed as the secondary ion quantification method, with a “Min. reporter PIF” set to 0.75. The analysis parameters included a reporter mass tolerance of 0.002 Da and a peptide mass tolerance of 10 ppm. The false discovery rates (FDRs) for peptides and proteins were kept below 1%. Enzymatic digestion allowed up to 2 missed cleavages by trypsin. The peptides were required to consist of at least six amino acids, with a maximum molecular weight limit of 12,000 Da. Methionine oxidation and acetylation at the N-terminus were assigned as variable modifications. For proteomics analysis, Cys carbamidomethylation was specified as a fixed modification, whereas for redoxomics analysis, Cys carbamidomethylation was designated a dynamic modification, and a new dynamic modification, “redox-biotin” ( + 414.194 Da), was added.
Redoxomics data processingThe proteomics data used for normalizing the redoxomics data were processed as follows. The proteinGroups.txt files were processed via R (version 4.3.0). Contaminants and decoy proteins were removed. The sum abundance in each TMT channel was scaled by the sum abundance of the common reference to obtain a total protein ratio. This ratio was then used to normalize the ratio of redox modifications.
The redox-biotinSites.txt file, containing the redox modification data, was processed as follows. Peptides marked with “+“ for reverse or potential contaminants, as well as those labeled with “REV_“ and “CON_“, were removed. The intensity of each peptide was normalized to the total protein ratio to obtain the normalized intensity. The normalized intensity of each biotinylated peptide of each sample was divided by the normalized intensity of the same peptide in the common reference sample. Furthermore, biotinylated cysteine residues derived from SH, Sto, SSG, SOH, and SNO, with probabilities greater than 0.75, were retained.
Proteomic data processingFor proteomics analysis, the “peptides.txt” file was processed via R (version 4.3.0). Contaminants and decoy proteins were removed, and unique peptides were extracted. Only proteins with ≥ 2 unique peptides were retained. The signal intensities of distinct unique peptides corresponding to the same protein were summed to quantify the overall abundance of the protein. To reduce sample loading variations, the total protein intensities in each sample were normalized to the same level. Additionally, protein intensities were further normalized against the common reference in each TMT batch to eliminate batch effects. Finally, all batches of data were merged, zeros were replaced with “NA,” the data were transformed to a log2 scale, and proteins identified in fewer than 50% of the samples were excluded from continued analysis.
Metabolomic data processingRaw data from untargeted metabolomics were processed via TraceFinder (Thermo, CA) on the basis of a custom-built metabolite database. The identification parameters included a mass tolerance of 10 ppm for precursor ions and 15 ppm for fragment ions, along with a permissible retention time shift of up to 0.25 min for quantitation. Metabolites with CV values ≥ 0.3 across quality control replicates were excluded to ensure reliability. Metabolite abundance was normalized by dividing each value by the corresponding protein concentration in the sample. Metabolites detected in positive and negative ion modes were normalized separately, with each metabolite’s intensity divided by a correction factor (correction factor = total intensity of each sample/average total intensity of all samples). Finally, the metabolites detected in both modes were combined.
Stoichiometry analysisFor each Cys residue, the stoichiometry of modifications such as SSG, SOH, SNO, or Sto was calculated by dividing the intensity of each modification by the sum of the intensities of Sto and SH. Cys residues quantified in at least three samples per group were retained, and the median occupancy of each modification on each Cys residue across different groups was used to generate the density distribution map.
Motif analysisMotif analysis was based on cysteine (Cys) residues located within ±10% of the Sto occupancy peak in both young and old individuals. Peptide sequences comprising 10 amino acids both before and after the identified modification site, totaling 21 amino acids, were selected for analysis. The analysis was conducted using the ‘ggseqlogo’ package in R (version 4.3.0).
Bioinformatics and statistical analysisThe proteomic, transcriptomic, phosphoproteomic, and metabolite data of the Macaca fascicularis colon tissues were normally distributed. To analyze differences in metabolite levels across three age groups, one-way ANOVA was used.
Differentially expressed redox modifications were screened via the following criteria. First, if the modification sites were detected in at least 3 samples in each age group, the disparities among the three age groups were evaluated using one-way ANOVA. Additionally, modification sites detected in more than half of the biological samples in one or two age groups but not detected in other age groups were also considered differential sites. To determine the age-related trends of these modifications, the fold changes between different age groups were compared.
The redoxomic data from mouse colon tissues were log2 transformed to ensure consistency with a normal distribution. For modification sites detected in at least three samples in both the control and the experimental groups, the Student’s t-test was used to evaluate statistically significant changes (p value < 0.05 and fold change > 1.2). Additionally, modification sites detected in more than half of the samples in one group but not in the other group were also defined as significantly different sites.
Spearman’s correlation analysis was used to assess the correlation between the two sets of omics data. Proteins significantly correlated with Cys modifications were subjected to Gene Ontology biological process (GOBP) enrichment analysis via the DAVID database. Metabolites significantly correlated with Cys modifications were subjected to Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis via MetaboAnalyst. Additionally, for enrichment analysis of mRNAs significantly correlated with Cys modifications, gene set enrichment analysis (GSEA) utilizing the hallmark gene set was carried out. All data analyses and graphical representations were carried out via R (version 4.3.0) and GraphPad Prism 9.0.
Procedures for assessing the functions of Cys residuesFull-length cDNAs encoding human genes (HPX, PYGB, RBBP9) were obtained from Miaoling Biology. Genes with point mutations (HPX-C408S, PYGB-C496S, and RBBP9-C39S) were constructed via PCR-mediated mutagenesis with 2X Phanta Max Master Mix (P525-01, Vazyme). The cDNAs were subsequently cloned and inserted into pLV3-CMV-MCS-3×FLAG-CopGFP-Puro vectors.
HEK293T cells were cultured in Dulbecco’s modified Eagle’s medium, while NCM460 normal human colon mucosal epithelial cells were grown in the RPMI-1640 medium. Both media were supplemented with 10% fetal bovine serum (FBS) (FSP500, ExCell Bio) and 100 U/mL penicillin plus 100 μg/mL streptomycin (10,099-141 C, Gibco). The wild-type and mutant genes packaged by lentiviruses were used to infect NCM460 cell lines. After 48 h, a portion of the cells was collected, and the expression of the wild-type and mutant proteins was detected via Western blotting.
The protein lysates were separated by 10% SDS‒PAGE and subsequently transferred onto a polyvinylidene fluoride (PVDF) membrane with a pore size of 0.22 μm (ISEQ00010, Millipore). After blocking, the samples were incubated overnight at 4 °C with primary antibodies (diluted 1:5000). HRP-conjugated secondary antibodies (diluted 1:10,000) were then incubated for 1 h at room temperature. Primary antibodies against Flag (66008-4-Ig, Proteintech) and GAPDH (60004-1-Ig, Proteintech) were utilized. The band intensity was evaluated via ImageJ.
NCM460 cells were harvested from a 10 cm culture dish and reseeded in a black 96-well plate. After 24 h, 200 μM H2O2 (Sigma-Aldrich, 88597) was added to the medium, and the mixture was incubated for 2 h. At the conclusion of the treatment period, the H2O2-containing medium was replaced with a fresh culture medium. After an additional 24 h, the medium was removed, and the cells were then rinsed thoroughly three times with Dulbecco’s phosphate-buffered saline (DPBS). Intracellular ROS levels were quantified via DHE (Dihydroethidium, Applygen, C1300-2). The cells were incubated with 100 μL of serum-free cell culture medium containing 10 μM DHE at 37 °C for 60 min. After incubation, the cells were washed with 1x PBS, and 150 μL of fresh DPBS was added to each well. Fluorescence measurements were then obtained via a microplate fluorescence reader (Bio-Tek) at excitation and emission wavelengths of 535 nm and 610 nm, respectively. Intracellular ROS production was normalized to the number of cells. The slope of ROS production was calculated relative to the expression levels of the wild-type or mutant proteins.
ROS measurements in cells treated with metabolitesNCM460 cells were seeded in 96-well plates at a density of 4000 cells per well in RPMI-1640 culture medium (C11875500BT, Gibco) and incubated at 37 °C for 24 h. The cells were then divided into 3 groups: the control group, the H2O2-treated group, and the metabolite- and H2O2-treated groups. The metabolites were dissolved in DMSO (196055, MP Biomedicals), and the cells in the metabolite and H2O2-treated groups were cultured with serum-free medium containing metabolites for 6 h, whereas those in the control group and H2O2 group were cultured with serum-free medium. H2O2 (200 μM) was subsequently added to the H2O2-treated group and the metabolite- and H2O2-treated groups, and the cells were subsequently incubated for 2 h. Afterward, all groups of cells were incubated with fresh medium at 37 °C for an additional 24 h. Finally, the intracellular ROS levels were measured as described above. The following metabolites were evaluated: allantoin (05670, Sigma-Aldrich), N-acetyl-L-alanine (A4625, Sigma-Aldrich), fumaric acid (47910, Sigma-Aldrich), indolelactic acid (I157602, Aladdin), CMP-N-glycoloylneuraminate (BT-T15, Beijing Bette Renkang Biomedical Technology), N-acetylaspartylglutamic acid (S879667, Macklin), prasterone sulfate (D609795, Aladdin), taurochenodeoxycholic acid (T303865, Aladdin), thiamine (WKQ0788740, Sichuan Vicchi Biotechnology), l-ornithine (L413185, Aladdin), chenodeoxycholic acid glycine conjugate (G304255, Aladdin), and glycocholic acid (G131002, Aladdin).
Fumarate treatment of DSS-induced colitisC57BL/6 female mice (7-week-old) were fed for 7 days before the start of the treatment. After acclimatization, the mice were randomly divided into three groups. The DSS + FA group received a daily oral gavage of 20 mg/kg body weight fumarate dissolved in PBS, whereas the control (CON) and DSS groups were gavaged daily with an equivalent volume of PBS. During the colitis induction period (day 2 to day 9), the drinking water for the DSS + FA and DSS groups was replaced with a 3% DSS solution. The CON group received regular water. Each group consisted of four mice. Daily records of body weight and disease activity indices were obtained. At the end of the experiment, the mice were humanely euthanized via cervical dislocation, and colon tissues were promptly excised for length measurement to assess inflammation.
HistopathologyColon tissues were fixed in 4% paraformaldehyde, followed by embedding in paraffin. The samples were then sectioned into thin slices, each measuring 5 μm in thickness, and subsequently stained with hematoxylin‒eosin. The sections were imaged via an automated digital slide scanner (PANNORAMIC MIDI, 3DHISTECH) and evaluated with CaseViewer (RRID: SCR_017654). Histopathological scoring was performed by two independent observers in a double-blind manner. Previous specific scoring criteria were applied.32
Calorie restriction of miceOne week prior to the calorie restriction experiment, 20-month-old mice were divided into two groups and housed in separate cages. The daily food consumption of the mice in each cage was recorded. During the initial week of the experiment, both the control normal (CN) group and the calorie restriction (CR) group were fed once a day. The CN group received 90% of their daily food intake, whereas the CR group received 80%. All the food provided was consumed by the animals. Beginning from the second week onward, the CN group continued receiving 90% of their daily food intake, whereas the CR group received 60% of their daily food intake. This regimen was maintained throughout the experiment, which lasted for two months. The mice were subsequently euthanized, and the ascending and descending colons were quickly separated and immediately frozen in liquid nitrogen for redoxomics, metabolomics, and proteomics analyses.
留言 (0)