
記住我



The transcriptomic profiles of paediatric and adult AML patients were collected from publicly available omics datasets accessed via the National Cancer Institute Data Portal (https://portal.gdc.cancer.gov/). The TARGET dataset (Bolouri et al. 2018)was used for the paediatric AML cohort, whereas the TCGA (Ley et al. 2013)and BEAT1.0 (Tyner et al. 2018) datasets were used for the adult AML cohort. Table 1 below shows the number of cohorts with the corresponding average age at diagnosis for each dataset. Although high-throughput data structures and gene expression signatures can be challenging to analyse owing to features such as being nonlinear and multivariate and having large structures, they do have some distinct advantages. Transcriptomic expression datasets are reproducible, robust, and tailored for broad coverage (Claudia Manzoni, Demis et al. 2016). Additionally, the extensive availability of transcriptomic data across diverse cancer types allows for robust cross-validation and comprehensive integration of findings. This abundance facilitates consistent analysis pipelines and enhances the reliability of the results. While proteomics and other omics types are valuable and could complement this work, they currently lack the same level of coverage and accessibility across datasets, which limits their integration in studies of this scale, making them excellent candidates for analysis (Kopec et al. 2005).
Table 1 Parameters/Selection criteria for each datasetThe elements of the TP53 pathway, as defined by the KEGG database, were identified and extracted from each dataset. This included the expression profiles of the TP53 pathway genes. These expression profiles were then used to create three distinct datasets corresponding to the BEAT, TCGA, and TARGET AML sources. The TP53 pathway was specifically targeted in this study because of its well-established role in regulating apoptosis, DNA repair, and cell cycle progression—functions frequently disrupted in AML and with strong therapeutic relevance (Granowicz and Jonas 2022). Additionally, alterations in the TP53 pathway are correlated with adverse prognoses in adult AML patients, underscoring its potential as a therapeutic target (Prokocimer et al. 2017; Zhu et al. 2023; Vadakekolathu et al. 2020).
To ensure comparability and consistency across datasets, min–max normalization was applied to scale the expression values of each dataset to a range between 0 and 1. This normalization process reduced variability arising from differences in data collection platforms and preprocessing pipelines, ensuring that all three datasets were suitable for subsequent analysis and were used as predictors in a stepwise artificial neural network to identify genes closely interacting with the TP53 pathway. To provide a comprehensive view, expression trends across the datasets were visualized in a heatmap, as shown in Supplementary File 1, which illustrates the mean comparative expression patterns across datasets. The expression trend shows that, while TCGA and BEAT share more similarities in TP53 pathway gene expression (reflecting consistency as expected within adult AML), TARGET exhibits a distinct expression pattern expected since it is a paediatric dataset, and the two conditions are clinically distinct (Farrar et al. 2016). Additionally, the expression trend serves to assess technical consistency and bias across datasets. Despite differences in patient demographics, sampling methods, and data processing protocols, the observed expression patterns remain broadly comparable, indicating that the trends are robust and not significantly influenced by technical artifacts. This provides reassurance that the findings are not only biologically meaningful but also reliable across independent cohorts.
Stepwise neural networkTo augment the TP53 pathway, as is currently understood, a stepwise artificial neural network (ANN) approach was applied, which allowed us to generate an enriched set of genes identified as expansions to the pathway. This neural network methodology allows the expansion of gene sets by identifying additional genes that interact closely and contribute to pathways. This approach has been previously proven successful in discovering and identifying novel drivers and biomarkers that can be used as therapeutic targets (Lancashire et al. 2009; Zafeiris et al. 2018). The stepwise ANN method utilized for the datasets used in this study was first described by Lancashire et al. (2008) and has been demonstrated to be effective in identifying patterns within the data by determining the optimum inputs for categorizing a specific task on the basis of the predictive performance of each variable, in this case, the expression levels of each gene in the TP53 pathway. This approach is especially helpful for biological datasets because it allows us to determine the genes most likely to account for a dataset’s variance (Zafeiris et al. 2018; Dhondalay 2013).
Neural networks present distinct advantages over traditional network enrichment analysis by leveraging their ability to uncover complex, nonlinear relationships in gene networks that are difficult for traditional methods to detect. Traditional network enrichment analysis, such as the approach described by Alexeyenko et al., enhances the statistical overlap of gene sets by incorporating network topology, making it more powerful than conventional gene set enrichment analysis (Alexeyenko et al. 2012). However, neural networks extend these capabilities by dynamically modelling interactions and capturing intricate patterns in multidimensional datasets. Unlike traditional methods that rely on predefined functional groups and statistical overlap, neural networks adaptively learn from data to reveal novel connections and biological insights, even for genes not well annotated in existing databases. This flexibility allows neural networks to provide richer, more accurate characterizations of gene relationships by integrating data from diverse omics sources, such as gene expression and protein interaction networks, without being constrained by fixed pathways or modules. Furthermore, the ability of neural networks to efficiently process high-dimensional data overcomes the curse of dimensionality, enabling the discovery of hidden interactions with enhanced scalability and robustness. This makes neural networks a transformative tool for functional genomics, offering deeper biological insights than traditional network enrichment approaches do (Lancashire et al. 2008; Tong et al. 2014).
The stepwise ANN is composed of a multilayer perceptron (MLP) with a single hidden layer containing two hidden nodes and a backpropagation algorithm that adjusts the network weight by feeding the error back through the model. A Monte Carlo cross-validation (MCCV) approach was used to stratify the data between training, validation, and testing with a 60:20:20 split, with 50 iterations used to ensure true randomization. It also utilizes an early stopping mechanism that allows the algorithm to stop early if no further improvements can be made to the model, with a minimum window threshold of 1000 iterations without improvement out of 3000 maximum iterations, which controls for possible overfitting of the data. A learning rate of 0.1 and a momentum of 0.5 were used to control the BP algorithm, and the model’s initial weights were set between 1 and − 1. The predictive power of each gene was evaluated via the mean square at the end of the process (Dhondalay 2013). The genes with the highest predictive power identified during the stepwise process as well as the genes belonging to the TP53 pathway were further analysed via the swarm-based deep neural network analysis approach, which is an improvement over methods that have been used extensively in other studies (Zafeiris et al. 2018; Dhondalay 2013; Lemetre et al. 2010; Wagner et al. 2018; Barron et al. 2018; Zafeiris et al. 2017) to predict how a given network of elements, in this case, genes interact with each other.
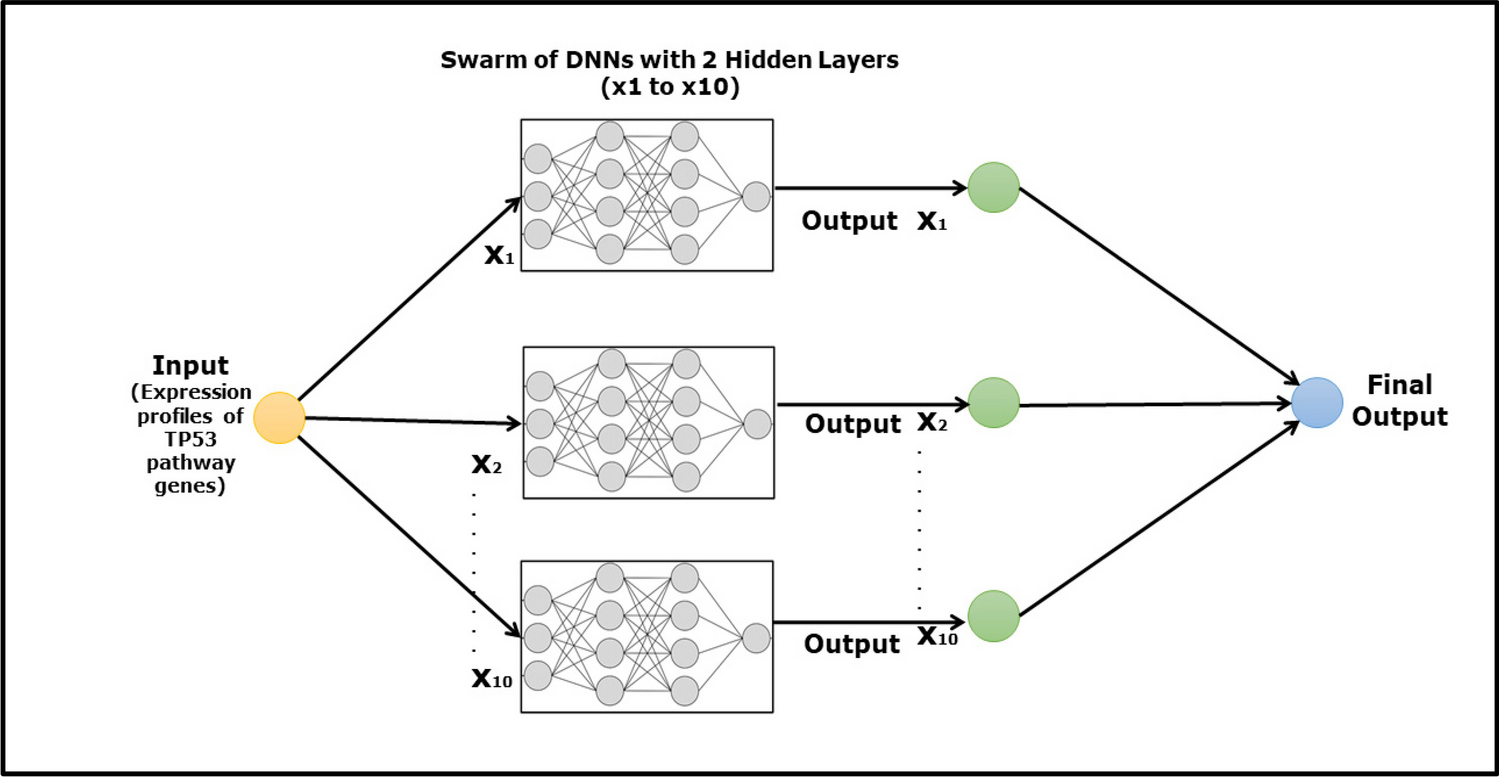
Swarm-based Deep Neural Network AnalysisArtificial neural networks mimic the function of human brain neurons by transmitting signals to connected neurons. They can recognize patterns and linearly separate them by giving each input a numerical weight value and adjusting it as the data are sampled, effectively finding the best possible solution to a specific question. A Swarm-based Deep Neural Network (SDNN) algorithm was employed to model the TP53 signalling pathway, and the architecture of the SDNN consisted of a three-layer feed-forward perceptron model, which was structured with the input layer containing the total number of genes (N), a hidden layer with two units, and a single output layer (N-2-1architecture) (see Fig. 1). A back-propagation algorithm was implemented to optimize the model, facilitating the identification of the most influential genes within the system. The algorithm allows for the determination of the central role within a system of the most influential genes, and the major advantage of this methodology is that it is multifactorial and evaluates each input, allowing the magnitude of the interaction for a particular parameter to be determined, whether it is inhibitory, stimulatory, unidirectional, or bidirectional (Lancashire et al. 2009; Dhondalay 2013; Lemetre et al. 2010). The Swarm-based deep learning model was developed by combining several smaller models, and the goal of this integration was to maintain interpretability, ensuring that the model’s depth of information and machine learning capabilities were preserved.
Fig. 1
Schematic illustration of the swarm-based deep neural network used in this study. Multiple DNNs labelled X1 to X10 operate in parallel to process an input. Each DNN receives the same input and independently generates an output. These individual outputs are then aggregated to produce a final, combined output. This approach leverages the collective processing power of the swarm of ANNs to increase the robustness and accuracy of the final output. The diagram shows the input flowing into each DNN and the subsequent flow of outputs converging into a singular final output, demonstrating the collaborative computation in the swarm-based model
The collaborative swarm for optimization or decision-making at each iteration offers significant benefits over traditional deep learning methods. The iterative process across multiple partitions increases adaptability and resilience, while leveraging swarm intelligence helps the model handle a wide range of scenarios, making it especially effective for dynamic and complex diseases such as cancer. This method also allows for exploring a broader solution space, potentially finding better configurations, and boosting overall model performance. Additionally, compared with traditional deep learning techniques, the collaborative swarm approach is significantly less computationally expensive; offers a high degree of biological explainability; and improves adaptability, robustness, and efficiency, making it a valuable strategy for challenging learning tasks (Zhang et al. 2021; Wu et al. 2011).
Learning was conducted over 3000 epochs with a termination factor of 1000 epochs, and the other parameters are listed in Table 2. The parameters listed in Table 2were carefully selected on the basis of prior optimization, testing, and validation performed by Tong et al. in 2014 (Tong et al. 2014), and Lemetre in 2010. These criteria were used to ensure the robustness and accuracy of the algorithm. A summary of their assessment results demonstrated high accuracy in terms of the true positive rate (TPR), correlation results, and interaction sign predictions, confirming the feasibility and reliability of these parameters. Notably, their findings revealed no significant improvement in the TPR when the number of hidden nodes increased, suggesting that the predictive ability of the algorithm is not significantly affected by this parameter. For example, this model with just two hidden nodes was found to perform better than models with more hidden nodes while requiring significantly less computational time. On the basis of these results, these parameters were implemented in the algorithm because they reflected the best performance for the time and cost of the analysis, optimizing both efficiency and effectiveness. In addition, the parameter configuration has been successfully validated and applied in multiple subsequent studies (Zafeiris et al. 2018; Lancashire et al. 2008; Tong et al. 2014; Zafeiris et al. 2017; Lemetre et al. 2009), demonstrating its robustness and reliability across diverse datasets and contexts. The optimization process ensures that the algorithm consistently delivers high predictive power under the given parameter settings, minimizing variability in the results.
Table 2 Interaction algorithm parametersAnalysing high-throughput omics data via deep learning models presents several challenges, including the high dimensionality of omics data, which can lead to overfitting and require advanced feature selection techniques. Deep learning models also lack the ability to make inductive predictions and determine the directionality of interactions (Lancashire et al. 2009). The stepwise artificial neural network approach addresses these challenges by incrementally adding layers and nodes to the network, managing complexity, and reducing overfitting risk. This method integrates feature selection within the training process, automatically identifying relevant features from omics data. Moreover, it enhances model interpretability by revealing key features for prediction. By iterative training on data subsets, the stepwise ANN approach optimizes performance with limited labelled data.
To mitigate the risk of overfitting, for both the stepwise ANN and the SDNN, independent transcriptomic datasets, specifically the TCGA, TARGET, and BEAT datasets, which are more recent, robust, and comprehensive, were employed. These datasets have provided robust validation for our findings and predictive models developed by the SDNN. Additionally, Monte Carlo cross-validation and an early stopping approach were employed. In addition, during training, the weights of the neural networks were regularized. Finally, the adopted concordance-based approach involving the use of multiple datasets, which is described in the differential driver analysis section, was a supplementary measure employed to mitigate the risk of overfitting.
Filtering and visualizing the interaction matrixA large matrix of interaction scores was generated by averaging the values across 10 iterations of the SDNN process until all the gene probes were used as outputs. The results were then sorted from largest to smallest with the initial values to obtain the distribution of the downregulated and upregulated interactions, followed by their absolute values to determine the strength of each interaction. The top 500 gene interactions based on the absolute interaction strength as well as interactions with SPAG5 specifically were then selected for model visualization via Cytoscape v3.10.2 (Shannon et al. 2003). Additionally, to reveal the significant module and hub genes closely interacting with SPAG5, MDM2, CDK1, and TP53, from their selection, the interactome models were analysed via MCODE (parameters: degree cut-off-4, node score cut-off-0.2, K-Core-2, and Max. depth-1000). Visualizing with Cytoscape makes it easier to create an interactive map with nodes representing sources and targets, edges representing relationships, the thickness of edges indicating the intensity of the association, different edge colours indicating whether the interaction is inhibitory or stimulatory, and arrows indicating the directionality of the pathway (Villaveces et al. 2015).
The approach further involved analysing an extensive interaction matrix generated from the network inference, which comprised ? = 83 genes. By considering each gene as an input, the model was designed to infer directed gene‒gene interactions/associations in a pairwise manner. The systematically formed associations resulted in n(n − 1) = 6,806 possible interactions, and this matrix enabled us to capture all potential gene‒gene relationships within the enriched TP53 pathway context. To specifically assess the interactions involving SPAG5, the interaction matrix was filtered to isolate associations with SPAG5. This filtering was applied to the interaction matrix generated from the three datasets—TCGA, TARGET, and BEAT—to ensure a robust and comparative analysis of SPAG5 interactions with key therapeutic targets. The absolute values of the interaction strengths were then employed to evaluate the robustness and potential biological relevance of each association, with higher values indicating stronger interactions.
Changes in the strength of interactions between key genes can significantly influence the stability and adaptability of the gene regulatory network. Stronger interactions may intensify regulatory control, potentially reinforcing feedback loops and stabilizing specific cellular states, such as cell cycle arrest or apoptosis, which are crucial in controlling cancer progression. Conversely, weakened interactions might reduce regulatory influence, destabilizing the network and allowing for unchecked cell proliferation or impaired apoptotic responses. These shifts could lead to network reorganization, with alternative pathways or compensatory mechanisms emerging to maintain functionality, though not always effectively. Understanding these dynamics is essential, as it enables us to predict how the network might respond to therapeutic interventions and to identify potential vulnerabilities that can be targeted to disrupt oncogenic processes (Li and Wang 2014; Rizi et al. 2021). These dynamics can be exploited by targeting key regulatory hubs with therapeutics that either weaken oncogenic interactions, destabilising cancer-promoting networks, or strengthen tumour-suppressing networks to restore normal function. Additionally, identifying compensatory mechanisms can guide combination therapies to prevent adaptive resistance and enhance treatment efficacy (Carels et al. 2023).
Differential driver analysisTo expand the understanding of the molecular pathways underlying cancer initiation and progression, transcriptomic-based data need to define and prioritize cancer drivers (Newberg et al. 2018)but one of the greater challenges is deciding upon the selection criteria that will allow for the elucidation of a driver or therapeutic target. Therefore, by adding the sum of weights that every source gene confers on each target and vice versa, it becomes possible to rank our candidates by their overall impact on the network as drivers, defined by the overall impact they have on other genes, and as receivers, defined by the overall impact other genes have on them. The SDNN process enables iterative quantification of the influence that various genes could have on the level of gene expression until all the genes in the data have been quantified, which enables us to develop a set of low-bias criteria determined by the impact each gene has on the network, the degree of influence source genes have on targets and the degree of influence a target has imparted on by source genes (Lemetre et al. 2010). The advantage of this method is that it is not biased towards single strong interactions, as it is simply the overall impact of each gene on the network allowing for the discovery of drivers with a relatively smaller but consistent impact on the overall network, which might otherwise be hidden by the selection criteria. These genes, while not obvious drivers, are likely key components of the disease system, and analysing them can provide us with a more comprehensive and unbiased view of the disease.
The methodology used in previous studies was then expanded with differential driver analysis, a comparative analysis that allows for the assessment of each potential driver’s impact and performance across multiple distinct patient groups. For the adult cohort, which included two datasets, the mean amount of influence for each gene was calculated and compared to that of the paediatric group. Differential driver analysis enhances the identification of key regulatory genes in different AML cohorts by allowing the assessment of each potential driver’s impact and performance across multiple distinct patient groups. For example, in the adult cohort, which included two datasets, the mean amount of influence for each gene was calculated and compared to that of the paediatric group. This comparative analysis helps identify genes that may drive AML pathogenesis in specific age groups, providing insights into age-specific differences in disease mechanisms. By calculating the mean amount of influence for each gene in each age group and comparing these values, researchers can identify genes that are more influential in driving AML in one age group than in another. This analysis can reveal the age-specific molecular mechanisms underlying AML and help identify potential therapeutic targets that are specific to certain age groups.
留言 (0)