
記住我

CBL proteins are a family of RING finger E3 ubiquitin ligases that regulate proliferative signals via ubiquitination and degradation of tyrosine-phosphorylated signaling proteins, such as KIT, CSF-1, FLT3 and PDGF [1]. We previously described c-CBL RING finger mutations in myeloid neoplasia (MN), chiefly myelodysplastic/myeloproliferative (MDS/MPN) syndromes such as chronic myelomonocytic leukemia [2, 3] and juvenile myelomonocytic leukemia [4]. c-CBL mutations are mainly heterozygous, but in some instances homozygous configuration can be found due to somatic uniparental disomy (UPD) affecting 11q chromosome [3]. Canonical c-CBL hits [missense (MS) mutations affecting linker and zinc finger domains] impair the ubiquitination function while preserving PI3K/AKT activation, common to RAS pathway, via LYN and KYN-mediated phosphorylation of c-CBL Y700, Y731, Y774.
Somatic c-CBL hits have been linked to monocytosis and MPN phenotype [5] and, across MDS and acute myeloid leukemia (AML) phenotypes, these hits cluster with other RAS pathway gene mutations in unsupervised molecular clustering schemes [6, 7]. Furthermore, outcome studies and prognostic scoring systems assigned a negligible prognostic role to c-CBL mutations in MDS [8] and AML [9], albeit without differentiating the type or mutational configuration. In addition to canonical mutations, c-CBL truncations can be also encountered and, since these lesions could impair not only the ubiquitin-association function, but also the PI3K/AKT activation function (enhanced through MS mutations), we hypothesized that these hits could lead to different, possibly less aggressive clinical phenotypes.
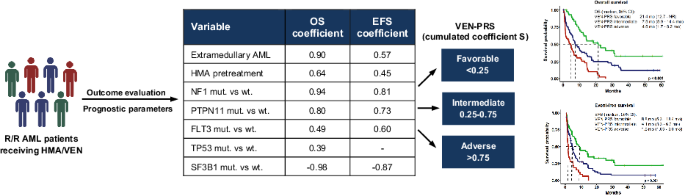
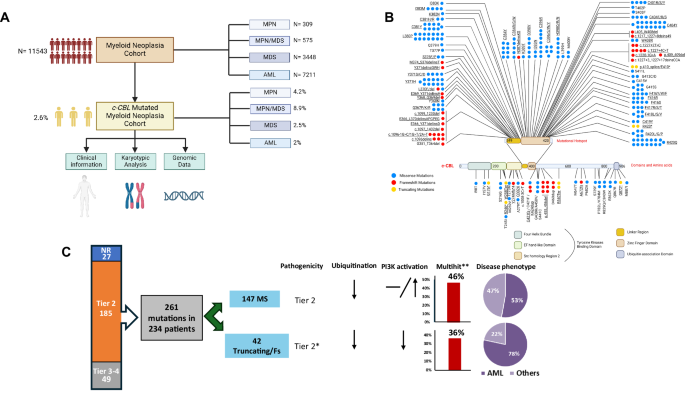
To test our theory, we identified several cases of canonical (pathogenic MS mutations occurring in the linker region/zinc finger domains) and truncating c-CBL mutations taking advantage of a large genotyped and clinically annotated cohort (n = 11,543: 7211 AML, 3448 MDS, 575 MPN/MDS; 309 MPN) (Fig. 1A, B, Supplementary Table 1). Clonal hierarchy of c-CBL mutations was assessed using the previously established variant allele frequency (VAF)-based algorithm [10]. Pathogenicity of the variants was assessed according to in-silico predictions tools and literature evidence allowing for classification into evidentiary tiers [11]. All the analyses were performed with tier 1 and 2 hits but, to prove our point, truncating [not in-frame indel, nonsense, splice-site; (T)] and frameshift (FS) mutations not previously described (19 variants) were considered as tier 2 and thus, were included as well (Fig. 1C).
Fig. 1: Characteristics of our cohort of c-CBL mutated patients.
A Flowchart of the study design and population selection. B Mutations distribution across the protein structure. Blue dots refer to missense mutations (MS), red dots to frameshift mutations (FS), yellow dots to truncating (T) (not in-frame indel, nonsense and splice-site) mutations. Underlined are the mutations further included in the analysis based on their pathogenicity. C Mutations selection. Mutations were divided according to their pathogenicity, functions (presumed from functional studies), and disease phenotype. We originally identified 322 mutations in 296 patients. Of these, the precise configuration was available in 261 cases (identified in 234 patients). According to our flow-chart, we selected, among MS variants, only tier 1–2 mutations occurring in linker region/zinc finger domains, and among T/FS variants, tier 1–2 mutations or mutations not previously described or with undefined pathogenicity, for a total of 203 variants in 184 patients: 147 harboring MS, 42 T/FS; 5 harboring both the type of mutation. More specifically, we included in MS cohort, 160 tier 1–2 mutations occurring in linker region/zinc finger domains (6 co-occurring with T/FS mutations) and in 21 T/FS tier 1–2 variants (2 co-occurring with canonical mutations) and 22 mutations without pathogenicity prediction (3 co-occurring with MS mutations). AML acute myeloid leukemia, FS frameshift, T truncating. *For the purposes of the analysis and based on literature reports we assumed that T/FS mutations not previously reported would be categorized as tier 2. **Mutations were considered multihits in case of two somatic lesions, VAF > 50%, del(11q) and/or chromosome 11 uniparental disomy.
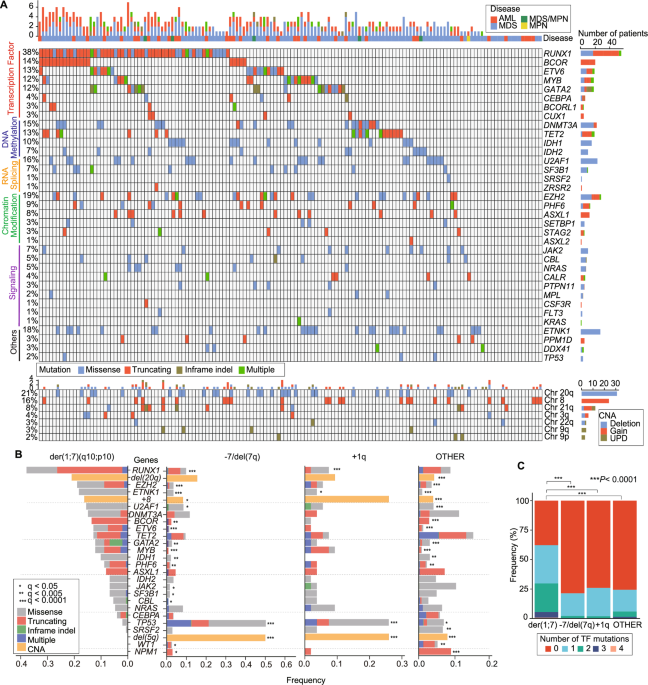
We identified 322 c-CBL mutations in 296 patients (Supplementary Table 2). Of these, the precise configuration was available for 261 c-CBL mutations, detected in 234 patients. According to our inclusion criteria, we focused on the variants of 184 patients harboring 203 tier 1–2 mutations (132 harboring one MS, 8 two MS, 2 three MS mutations; 36 harbored one T/FS, 1 two T/FS, 4 MS and T/FS, 1 with two MS and 1 harboring T/FS mutations) and their clinical, mutational, and cytogenetic characteristics were analyzed (Fig. 1A). In agreement with previous studies, most of the mutations occurred in “hotspot regions” (linker and zinc finger domains; 100% of MS mutations, according to our selection criteria, and 67% of T/FS) (Fig. 1B). Biallelic hits, defined by: (i) combined VAF > 50% of 2 distinct c-CBL mutations, (ii) somatic UPD or microdeletion involving mutant c-CBL locus (1 mutation with VAF > 50%), (iii) del(11q) concomitant with c-CBL mutation, were found in 46% in MS group and 36% in T/FS group (p = 0.22; Fig. 1C).
There were significant differences in the distribution of c-CBL T/FS and MS mutations among the disease phenotypes: in the former group we observed 29 AML (78%), 2 MDS (5%), 4 MDS/MPN (11%), 2 MPN (5%); in the latter 75 AML (53%), 21 MDS (15%), 36 MDS/MPN (25%), 10 MPN (7%), suggestive of the association of T/FS mutations with more aggressive clinical phenotypes. In fact, T/FS vs. canonical MS mutations were more common in AML in general (78% vs. 53%, p = 0.005) and primary AML (pAML, 90% vs. 68%, p = 0.023) (Fig. 1C, 2A, Supplementary Table 3).
Fig. 2: c-CBL mutations comparison.
For the analysis, we divided c-CBL in missense (in black) and truncating/frameshift (in gray) mutations. A Disease phenotype. Truncating/frameshift mutations showed higher frequency in AML phenotype and in primary AML. For consistency, we focused only on AML in subsequent comparisons. B Karyotype features. C Mutational landscape. D Multivariate analysis of overall survival. Forest plot of hazard ratio of the variables included in the model whit 95% confidence intervals. For more details refer to Supplementary Table 4. Asterisk in the figure refers to statistically significant (P ≤ 0.05) correlations. AML acute myeloid leukemia, FS frameshift, MDS myelodysplastic syndromes, MPN myeloproliferative neoplasms, pAML primary AML, sAML secondary AML.
Considering the abundance of AML in T/FS group, we decided to focus our analysis on this phenotype. AML patients with T/FS mutations tended to harbor abnormal karyotype compared to patients with MS mutations (31% vs. 53%, p = 0.09), had higher percentage of complex karyotype (19% vs. 4%, p = 0.016, Fig. 2B), lower percentage of co-mutations in splicing factor genes (in particular SRSF2: 7% vs. 27%, p = 0.025) and showed a trend to co-occur with del(17p) (8% vs. 1%, p = 0.10) and TP53 mutations (10% vs. 3%, p = 0.10, Fig. 2C). However, no differences were detected in clonal hierarchy in both the subgroups wherein c-CBL mutations were frequently sub-clonal (T/FS 63% vs. MS 47%, p = 0.16). In T/FS cohort hierarchical analysis was conducted in 27 cases: 7 hits (26%) were ancestral, 1 (4%) the only mutation detected, 2 (7%) codominant, 17 (63%) sub-clonal. In the MS cohort, hierarchical analysis was conducted in 72 cases: 21 hits (29%) were ancestral, 4 (5%) the only mutations detected, 13 (18%) codominant, 34 (47%) sub-clonal. In terms of ELN risk stratification [9], we observed an enrichment in high-risk AML in both T/FS (50%) and MS mutations (62%) (Supplementary Fig. 1A, B). Co-mutational landscape of the two subgroups showed a positive correlation between MS mutations and SRSF2 [odds ratio (OR):4.5, 95% confidence intervals (CI):0.981–20.634; p = 0.05], and inverse correlations between MS mutations and TP53 (OR:0.160, 95% CI:0.028–0.928; p = 0.02) and T/FS mutations and SRSF2 (OR:0.175, 95% CI:0.038–0.784; p = 0.011) (Supplementary Fig. 1C). When we conducted survival analysis, AML patients with T/FS mutations presented a trend toward worse prognosis [median overall survival 11 vs. 16 months of AML patients with canonical RING finger MS mutations, p = 0.161; vs. 14.3 months of AML patients with wild type (wt) c-CBL, p = 0.20 (Supplementary Fig. 2)] but this difference was not significant. However, Cox regression model showed an independent role of T/FS mutations as risk factor for poor outcome (Hazard ratio:1.642, 95% CI:1.002–2.690; p = 0.049; Fig. 2D, Supplementary Table 4). Of note is that the favorable outcome of MS mutations was not confirmed in the multiple regression model (Supplementary Table 5). Biallelic status and hierarchical status of mutations did not show a different prognosis in MS or in T/FS group (Supplementary Figs. 3 and 4). When we analyzed the features of 5 patients harboring both MS and T/FS mutations, all of them had pAML with normal karyotype, c-CBL mutations were co-dominant/subclonal in 4/5 cases but showed a diverse mutational spectrum (Supplementary Table 6). There was also no clear trend towards dominant expansion between T/FS and MS mutations occurring in in the same patient. These findings suggest that the co-existence of different type of c-CBL mutations could produce mixed MN phenotypes without a clear hierarchical predominance.
Finally, given the prognostic weight of c-CBL mutations, we investigated a potential role also in less-acute phenotypes. We stratified MDS and MDS/MPN patients according to IPSS-M score. However, our cohort includes a paucity of MDS and MDS/MPN cases harboring T/FS hits (n = 6), which precluded qualified analysis. When we focused on canonical MS mutations, we were able to classify 44 patients [6 (14%) low risk, 7 (16%) moderate low, 7 (16%) moderate high, 13 (29%) high, 11 (25%) very high] while in T/FS cohort we classified only 4 patients [1 low risk, 1 moderate low risk, 1 high risk, 1 very high risk]. Thus, no IPSS-M risk class presented enrichments in c-CBL mutated population. The survival analysis of patients with MS showed, as expected, a better outcome in lower-risk categories (p = 0.02; Supplementary Fig. 5).
Our study highlights the importance of distinction of “non canonical” c-CBL mutations and T/FS variants whose impact could be underestimated and suggests that AML harboring different types of c-CBL mutations present profound differences in disease phenotype, co-mutational landscape, and outcome.
Strikingly, T/FS mutations occur often in pAML and convey a less favorable prognosis, independently from the co-occurrence of high-risk features, such as complex karyotype, del(17p) and TP53 mutation. Both these findings could indicate a more aggressive behavior of T/FS hits, causing a faster transition from MDS/MPN to AML (which, thus, were classified as pAML) and a dismal outcome. This feature seems to rely on leukemogenic pathways other than RAS signaling, as also suggested by a recently reported case of histiocytic neoplasm showing resistance to MEK1/2 inhibition after acquisition of a T/FS c-CBL mutation [12].
In this perspective, the virulent nature of T/FS variants, not explained by RAS pathway activation, could stem from different mechanisms, such as a dominant-negative effect of T/FS hits on wt c-CBL preventing ubiquitination, as shown in solid tumors [13] and suggested, in MN, by studies demonstrating not only a dismal prognosis in c-Cbl knock out mice when compared to wt counterparts, but also an enhanced proliferative response to cytokines [14].
Interestingly, the hierarchical analysis of c-CBL mutations, which showed that, roughly, in 1/3 of cases (30% T/FS, 35% MS mutations) c-CBL is the only mutation detected or the founder lesion, suggests that both types of c-CBL hits could represent primary leukemogenic drivers, arguing against previous observations [15].
However, it should be highlighted that VAF method based on NGS to distinguish ancestral/founder hits has various shortcomings because clones affected by different mutations may result in the growth arrest and thereby lower VAF values. This is likely due to the contribution of wt DNA from mature myeloid cells produced by residual normal hematopoiesis. Nevertheless, the survival analysis according to the crude hierarchical assignment of c-CBL mutations by clinically easily applicable VAF methods indicates that c-CBL mutations, irrespective of their position in the clonal architecture or timing of occurrence, have similar impact to disease evolution.
We conclude that divergent impact on leukemogenesis of MS vs. T/FS c-CBL mutations leads to a differential impairment of the protein function and might be responsible for the different MN phenotypes and outcomes observed.
留言 (0)