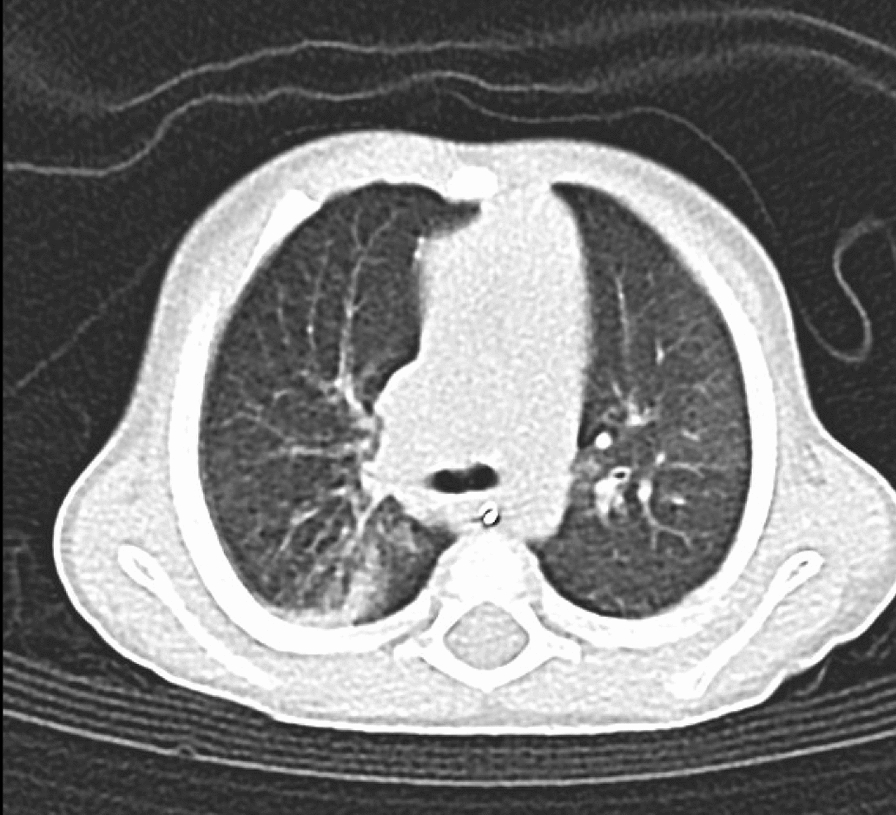
This clinical presentation is notable for the severe and recurrent infections, growth restriction, adrenal insufficiency, and enteropathy, among other complications. This aligns with the emerging understanding of MIRAGE syndrome as a complex multisystem disorder with diverse and often severe clinical features. Some of those features may be more unique in our case and will be discussed along the following lines.
To our knowledge, there are no reported cases of MIRAGE syndrome associated with pyloric stenosis. Two previous cases of adrenal insufficiency associated with pyloric stenosis were reported, but neither had a syndromic cause [9]. Theoretically, pyloric stenosis is unlikely in MIRAGE syndrome given that MIRAGE syndrome mostly manifests by growth suppression of organs rather than hypertrophy, which is the underlying pathology in pyloric stenosis [10]. Reported GI manifestations of MIRAGE syndrome include achalasia, gastroesophageal reflux and recurrent intussusception [3]. While other factors can contribute to pyloric stenosis such as medications, no specific predisposing factor was found in our case. CNV 601 259 bp duplication is established to cause hypertrophy in structures like the heart, but no evidence of its relation to the pylorus [11]. Therefore, it is only a theoretical manifestation and further studies are needed. Moreover, whether or not the manifestation of pyloric stenosis represents a possible phenotype of this variant of MIRAGE syndrome is an association yet to be investigated.
The varieties in the manifestations of MIRAGE syndrome can overlap with other syndromes, which should be included in the differential diagnosis prior to genetic testing. For example, the clinical presentation of our patient can be compared to the typical presentation of IMAGe (Intrauterine growth restriction, Metaphyseal dysplasia, Adrenal hypoplasia, Genital anomalies) syndrome. While both syndromes involve adrenal hypoplasia, they exhibit distinct features [12]. IMAGe syndrome is characterized by intrauterine growth restriction, metaphyseal dysplasia, and adrenal hypoplasia. In contrast, MIRAGE syndrome, as exemplified by our case, is more phenotypically diverse, encompassing myelodysplasia, infection, growth restriction, adrenal hypoplasia, genital anomalies, and enteropathy. The phenotypic diversity of MIRAGE syndrome is evident in the variable or milder features observed in some individuals, in contrast to the initially reported severe presentations. [4] However, there is still lack of functional validation of the variant identified. Furthermore, the role of CNV duplication in this case also requires further validation.
The genetic basis of the two syndromes also differs. IMAGe syndrome is associated with mutations in the CDKN1C gene [13], while MIRAGE syndrome is linked to heterozygous activating mutations in the SAMD9 gene [4]. Furthermore, the clinical implications and management of the two syndromes vary. IMAGe syndrome is primarily characterized by prenatal and postnatal growth restriction, skeletal abnormalities, and adrenal insufficiency, with a focus on endocrine and skeletal assessments. On the other hand, MIRAGE syndrome presents a more complex and multisystem involvement, necessitating a comprehensive approach to address the diverse clinical features, including myelodysplasia, infections, and enteropathy, in addition to growth restriction and adrenal hypoplasia.
X-linked adrenal hypoplasia congenital syndrome, which is an X-linked inheritance pattern syndrome that affects NROB1 (Xp21.1–21.2) gene, can also be a mimic of MIRAGE syndrome which may be considered in its differential. While both syndromes share the similarity of adrenocortical hypoplasia, X-linked adrenal hypoplasia congenital syndrome shows other clinical features which differs from that of MIRAGE syndrome, which include hypogonadotropic hypogonadism, delayed puberty, and azoospermia.
Another syndrome that may resemble the presentation of MIRAGE syndrome is Familial glucocorticoid deficiency, which is an autosomal recessive disease that affects the MC2R & MRAP genes (18p11 & 21q22, respectively). Familial glucocorticoid deficiency shares 2 main characteristics with MIRAGE syndrome which are recurrent infections and adrenocortical hypoplasia. However, familial glucocorticoid deficiency is unique to its other clinical features that include hyperpigmentation, failure to thrive, early onset of hypoglycemic seizures, and even collapse or coma [14]. Another disease that is getting more mistakenly considered as MIRAGE syndrome is familial dysautonomia (FD) which is a rare genetic disease that is caused by a variant in the ELP1 gene. FD shares some clinical features of MIRAGE syndrome such as growth restriction, hypotonia, frequent lung infection, and feeding difficulties. As a result of these similarities constructing the diagnosis of MIRAGE syndrome can be a bit more challenging process [15]. All of these syndromes show how structural variations can contribute to multisystemic disorders.
Clinical manifestations and phenotypic expression of MIRAGE syndrome can be related to SAMD9 variations [3]. Interestingly, in patients with gain of function mutation of the SAMD9, secondary events may occur to decrease the effects of growth suppression, such as loss of function mutation of SAMD9 [3]. This is especially true for hematopoietic cells, and this contributes to the diverse phenotypes. Moreover, some pathological variants were found in patients with hypospadias born with small weight without adrenal insufficiency, while other patients demonstrated thyroid gland disease [3]. Finally, patients with isolated myelodysplastic syndrome also demonstrated variations in SAMD9 gene. However, MIRAGE-associated variants are clustered around P-loop NTPase domain, while isolated MDS variants are present across protein domain structure [3]. To our knowledge, no specific report discussed the variant Pro295GlnfsTer104. Moreover, no previous research was found on cases with both SAMD9 mutation and CNV-related duplication which our patient had. Further research might contribute to better understanding of these two entities. A summary of existing literature description of MIRAGE syndrome phenotypes and genotypes may be found in Table 1. Due to the nature of our study, limitations include lack of generalizability, retrospective design, lack of ability to establish a cause–effect relationship, and potential information bias from subjective interpretation.
Table 1 Phenotypic and genotypic variations of MIRAGE syndrome in existing literature related to SAMD9 mutationThe identification of pyloric stenosis highlights the importance of considering a broader gastrointestinal manifestations that could complicate the course of the illness. A comprehensive evaluation and multidisciplinary management are essential due to the heterogeneity of the clinical presentations. Furthermore, understanding the interplay between SAMD9 mutations and other genetic alternations, such as CNV duplications, may guide personalized approaches to management. Further research is needed to explore the genetic and phenotypic variability of MIRAGE syndrome and the mechanisms underlying organ-specific manifestations, which could provide insight into targeted interventions. Case series or longitudinal studies with larger samples could highlight potential causal associations.





留言 (0)