
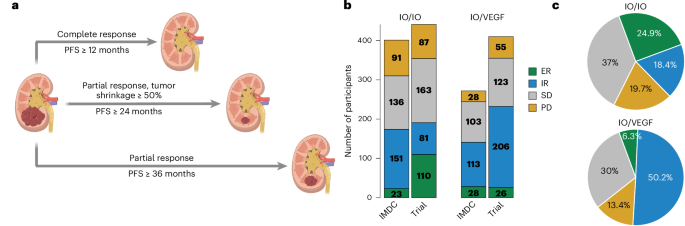
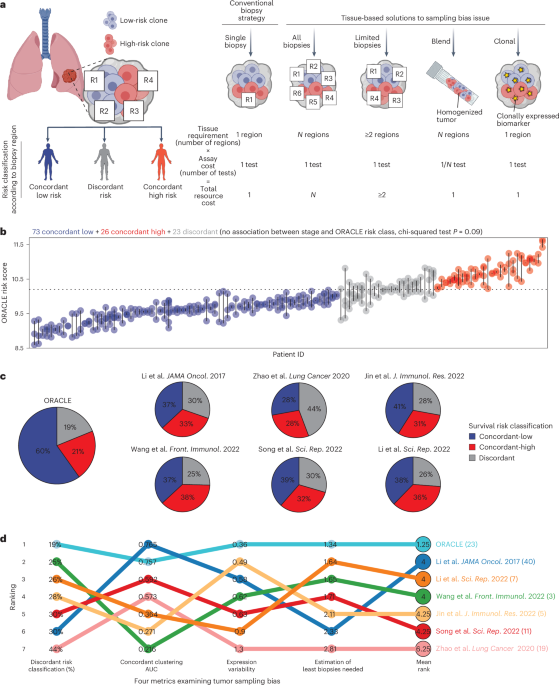
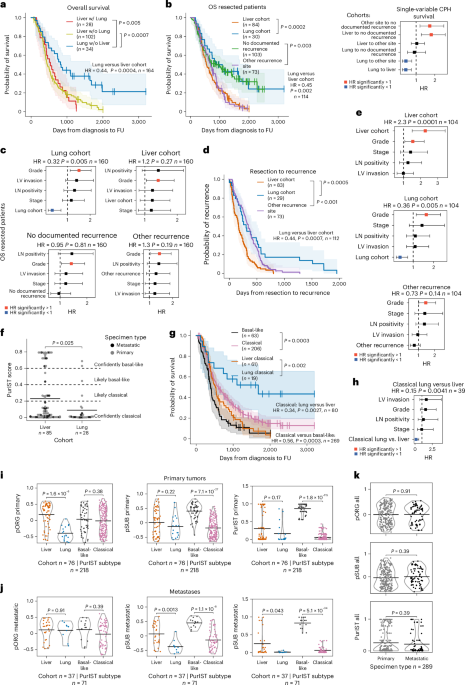
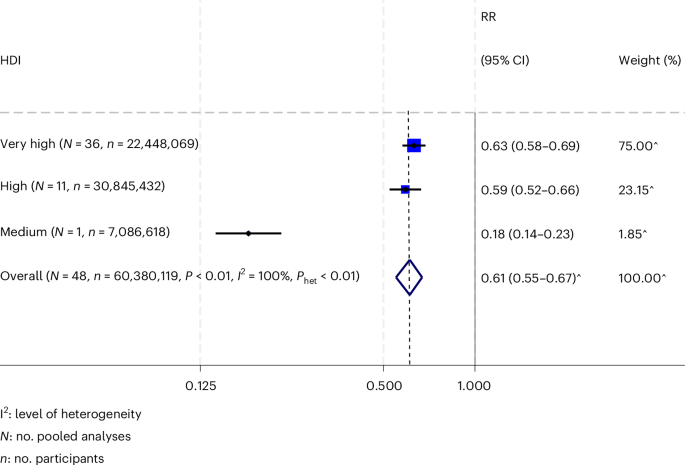
記住我
All research performed in this study complied with all relevant German ethical regulations.
Clinical specimensLiquid biopsy samples (pleural and ascitic effusions and peripheral blood withdrawals) were obtained from MBCPs participating in the CATCH (Comprehensive Assessment of Clinical Features and Biomarkers to Identify Patients with Advanced or Metastatic Breast Cancer for Marker Driven Trials in Humans) trial at the Division of Gynecologic Oncology, NCT Heidelberg (case number S-164/2017). Written informed consent was obtained from all participants.
Participant and tumor characteristics are summarized in Supplementary Table 1. CTC-specific assessments were further approved by the ethical committee of the University of Heidelberg (case number S295/2009) and University of Mannheim (2010-024238-46).
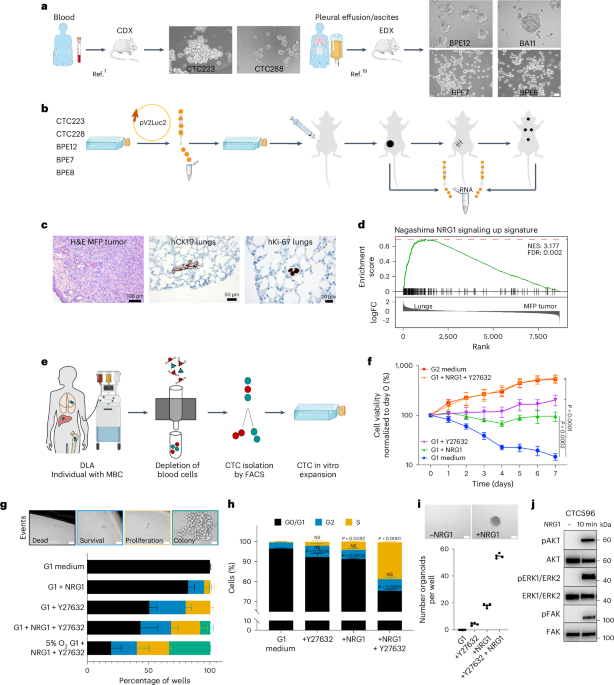
EDX and CDX modelsThe procedure for generating orthotopic xenografts from effusion was first described by Al-Hajj et al.43 and was recently refined by our group19; peripheral blood withdrawal was previously described by our group1.
CTC enumerationBlood samples were collected in CellSave tubes (Menarini Silicon Biosystems) and run with CellSearch (Menarini) using the CTC kit (Menarini) at the Department of Tumor Biology, Hamburg, according to the manufacturer’s guidelines.
Blood samplesPeripheral blood samples were directly collected in Vacutainer CPT tubes (BD Biosciences) and processed following the manufacturer’s guidelines.
LeukapheresatesMBCPs with ≥10 CTCs per 7.5 ml of blood were asked to participate in the CTC leukapheresis study, approved by the ethical committee of the University of Heidelberg (case number S-408/2013). DLA was performed at the Medical University Hospital Heidelberg using a Spectra Optia (Terumo BCT) according to manufacturer’s instructions. Plasma and CTCs were collected using the cMNC program with a reduced packing factor of 4.5 and with 2–3% hematocrit.
Apheresate products were immediately collected and processed under sterile conditions. Depletion of blood cells was performed using Miltenyi Biotec microbeads (anti-CD45, 130-045-801; anti-CD3, 130-050-101; anti-CD31, 130-091-935; anti-CD16, 130-045-701; anti-CD235a, 130-050-501; each 20 µl per 107 cells).
FACS and flow cytometryCells were stained in PBS + 1% bovine serum albumin (BSA) and 2 mM EDTA using the following antibodies: EpCAM-FITC and APC-Vio770 (clone HEA-125, REA764 Miltenyi Biotec, 1:50), CD45-VioBlue (REA747 clone, Miltenyi Biotec, 1:50), HER3-PE (66223 clone, FAB3481P, R&D, 1:20), CD31-VioBlue (clone AC128, Miltenyi Biotec, 1:50), CD16-VioBlue (clone REA423, Miltenyi Biotec, 1:50), CD41-VioBlue (clone REA386, Miltenyi Biotec, 1:50) and CD235a-VioBlue (clone REA175, Miltenyi Biotec, 1:50). Propidium iodide (PI; P3566, Thermo Fisher Scientific, 1:1,000) or DAPI (D1306, Thermo Fisher Scientific, 1:1,000) was used to exclude dead cells. The following antibodies were used for the analysis of xenograft-derived samples (all anti-mouse, PacificBlue, Biolegend): CD45 clone 30-F11 (103116, 1:1,000), CD11b clone M1/70 (101226, 1:2,000), TER-119/Ly-76 clone TER-119 (116223, 1:200), Ly-6G clone 1A8 (A25985, 1:2,000), CD31 clone 390 (102422, 1:1,000) and H-2Kd clone SF1-1.1 (116629, 1:50).
Cells were sorted and analyzed using a FACSAria Fusion and LSR Fortessa (BD Biosciences). BD FACSDiva software and FlowJo software were used for analysis.
Determination of NRG1 blood concentrationNRG1 concentration in blood samples from MBCPs was measured with a Human NRG1 ELISA kit (Thermo Fisher Scientific) according to the manufacturer’s instructions. Absorbance was determined on a SpectraMax iD3 microplate reader.
Long-term in vitro expansion of CTCsImmediately after FACS or immunomagnetic sorting, the CTC-enriched cell suspension was centrifuged, the supernatant was carefully removed, and the pellet was resuspended in the appropriate volume of CTC medium, transferred to a plate (Corning, Primaria) and cultured at 37 °C with 5% CO2 and 5% O2. Medium was partially replaced every 48–72 h. When cluster formation was detected, cells were collected, dissociated with Accutase (Sigma, Life Technologies) and expanded in larger plates and subsequently in flasks. Floating three-dimensional collagen gels with a final concentration of 1.3 mg ml–1 collagen I (Corning) were prepared as previously described with minor modifications20. NRG1 concentration used in the medium and in all experiments was 20 ng ml–1 unless otherwise specified. The medium recipe was licensed to Miltenyi Biotec, and the optimized formulation is available under the name ‘Breast TumorMACS Medium’; only Y27632 has to be added separately according to the manufacturer’s instructions (StemMACS Y27632, 130-103-922).
The cells were confirmed to be negative for contaminants (no Mycoplasma, Squirrel Monkey Retrovirus, Epstein-Barr virus or interspecies contamination was detected) by the Multiplex Cell Contamination Test (Multiplexion)44. To test cross-contamination with commercially available cell lines, single-nucleotide polymorphism fingerprints were extracted from RNA-seq data on CDOs in vitro and publicly available RNA-seq data from BC and acute myeloid leukemia cell lines from CCLE45 (PRJNA523380) using ExtractFingerprints and CrosscheckFingerprints from GATK4 (ref. 46). Briefly, given a BAM/SAM/VCF file, single-nucleotide polymorphisms in specific highly variable genomic locations were extracted and calculated. Concordance between fingerprints was estimated, and the logarithm of the odds score for identity was calculated (Extended Data Fig. 4a).
The match between CDOs and corresponding participant material was confirmed by ExtractFingerprints and CrosscheckFingerprints from GATK4 on whole-genome sequencing data (Extended Data Fig. 4b).
Clonogenic assaySingle-cell suspensions were obtained by treating cells with Accutase at 37 °C for 5 min. Cells were washed with PBS, spun down, resuspended in a PBS solution containing 1% BSA, 2 mM EDTA and DAPI and filtered through a 40-µm cell strainer. Cells were sorted using a FACSAria Fusion (BD Biosciences) coupled with BD FACSDiva Software using a 100-μm nozzle. After morphological gating using forward and side scatter, we excluded duplets and dead cells (DAPI+) and sorted one single cell per each well of a 96-well plate, previously filled with 200 µl of medium. All plates were maintained at 37 °C with 5% CO2 and 20.9% O2 for normoxia and 5% O2 for hypoxia. After 2 or 4 weeks, cells were checked and counted under brightfield microscopy.
CRISPR–Cas9-mediated ERBB3 KOERBB3-KO gRNA sequences (Supplementary Table 1) were cloned into the pLKO.005 backbone of the pLKO5.sgRNA.EFS.tRFP657 plasmid, as previously described47,48. The correct assembly of the resulting DNA plasmids was confirmed using Sanger sequencing. Lentivirus production was performed using a second-generation lentivirus system (psPAX2, Addgene plasmid 12260; pMD2.G, Addgene plasmid 12259). Viral supernatant was collected 48 h after transfection, passed through a 450-nm filter, ultracentrifuged at 4 °C for 120 min, resuspended in cold PBS and stored at −80 °C until use. CTC223 cells were transduced with pCW-Cas9-tGFP plasmid. After 4 days of incubation, the GFP–Cas9+ CTC223 cells were sorted by FACS, expanded and transduced with pLK05-gRNA1-tRFP (ERBB3-KO1), pLK05-gRNA2-tRFP (ERBB3-NF) or pLK05-EV-tRFP (EV). GFP+tRFP+ cells were sorted by FACS and expanded. After 10 days, ERBB3 KO was induced by adding 1 μM doxycycline hydrochloride to the medium for 96 h and exchanging the medium after 48 h.
Cell viability assayCell viability was measured using the CTB (Promega) assay. Specifically, 8,000–10,000 cells were seeded under the appropriate medium conditions in each well of a 96-well multiwell plate. The day after, the following compounds were added: AZD4547 (S2801), lapatinib (S2111), taselisib (S7103) and alpelisib (S2814; all purchased from Selleckchem).
Cell cycle analysisIn total, 1 × 105 cells per well were seeded in G1 medium. After 24 h, the medium was replaced with G1, G2, G1 + NRG1, G1 + Y27632 and G1 + NRG1 + Y27632. After 48 h, 5-ethynyl-2′-deoxyuridine (EdU; 1:1,000) of a Click-iT Plus EdU Alexa Fluor 647 Flow Cytometry Assay kit (Invitrogen) was added. After 3 h, cells were processed according to the manufacturer’s instructions and analyzed using an LSR Fortessa.
Apoptosis assayIn total, 1 × 105 cells per well were seeded in G1, G2, G2 without NRG1 or G2 without Y27632. After 48 or 120 h, cells were washed with PBS and detached with Accutase. Cells were then stained in 1× Annexin V Binding buffer with 1:20 PI and 1:100 Annexin V-PE. After 15 min of incubation at room temperature, samples were measured using an LSR Fortessa. The fractions of alive (PI–Annexin–), early apoptotic (PI–Annexin+), late apoptotic (PI+Annexin+) and necrotic (PI+Annexin–) cells were determined using FlowJo software.
Synergism between NRG1 and Y27632To investigate the synergistic effect of NRG1 and Y27632 on CTC proliferation, we used a linear model to describe cell viability. This model included binary variables for NRG1 treatment, Y27632 treatment and their combination. To test for synergy, we compared two generalized linear mixed models: one with and one without an interaction term for the double treatment. An ANOVA revealed a significant improvement in the model with the interaction term (P = 1.25 × 10–6), indicating a synergistic effect.
Mouse studiesAnimal care and procedures followed German legal regulations and were previously approved by the governmental review board of the state of Baden-Württemberg, operated by the local Animal Welfare Office (Regierungspräsidium Karlsruhe) under license numbers G-240/11, G-115/17 and G-104/22. Mice were housed in individually ventilated cages under temperature and humidity control. Cages contained an enriched environment with bedding material.
In vivo preclinical model for micrometastasesLuciferase-labeled CDX models were injected into the fourth mammary fat pad of female NSG mice that were at least 6 weeks old. Cells were resuspended in a 1:1 ratio of sterile PBS and growth factor-reduced Matrigel (BD). All mice received subcutaneous implantation of β-estradiol as solid pellets (Innovative Research of America), as previously described1. Primary tumors were resected after reaching a size of ~0.5 cm3. Micromestastasis formation was monitored by measuring bioluminescent signal using an IVIS Spectrum Xenogen device (Caliper Life Sciences). Animals were killed, and micrometastasis-containing organs were collected.
CDX generationNSG mice were transplanted with 1 × 106 cells in the fourth mammary fat pad, as described above. Tumor growth was monitored, and mice were killed when end point criteria were reached.
In vivo lung colonization assayIn total, 1 × 105 cells in 100 µl of PBS were injected via the tail vein. Lung colonization was monitored by measuring bioluminescent signal using an IVIS Spectrum Xenogen machine (Caliper Life Sciences). Bioluminescence analysis was performed using Living Image software version 4.4 (Caliper Life Sciences).
In vivo drug treatmentsTumor growth was monitored, and drug treatment was initiated when primary tumors were palpable. Mice were treated by oral gavage daily with vehicle (0.1% Tween 80/0.5% Na-CMC, 100 μl), 100 mg per kg (body weight) lapatinib, 10 mg per kg (body weight) AZD4547 or 100 mg per kg (body weight) lapatinib + 10 mg per kg (body weight) AZD4547 all in 0.1% Tween 80/0.5% Na-CMC (100 µl). Tumor size was recorded weekly using digital calipers. Animal weight was recorded daily to monitor potential drug toxicity. At the experimental end point, the maximal tumor size (1.5 cm3) was not exceeded. None of the treatments had major toxic effects in vivo (Extended Data Fig. 10b).
Gene expression analysis by quantitative PCRCells were collected, washed with PBS and centrifuged. RNA extraction was performed using miRNeasy Mini kit (Qiagen) or PicoPure kit (Thermo Fisher), depending on the cell number, following the manufacturers’ instructions. RNA concentration and purity were determined using a NanoDrop spectrophotometer. Reverse transcription (RT) was performed using a High-Capacity cDNA Reverse Transcription kit (Applied Biosciences) following the manufacturer’s guidelines. Quantitative PCR was performed using 0.5 μl of forward and reverse primers (10 μM stock solution), 5 μl of Power SYBR Green PCR master mix (Life Technologies), 3 μl of nuclease-free water and 1 μl of 1:10 diluted cDNA. The reaction was run on a Thermo Fisher ViiA-7 Real-time PCR system. Data were analyzed using the comparative cycling threshold (ΔΔCt) method. Primers are listed in Supplementary Table 5.
Gene expression analysis of the PDX modelPrimary tumor and metastatic tissues derived from our PDX mouse model were freshly processed using the GentleMACS system (Miltenyi Biotec) to obtain a single-cell suspension. Live tumor cells were sorted by FACS into RNA lysis buffer (Arcturus PicoPure RNA Isolation kit, Life Technologies, Invitrogen). RNA was isolated according to the manufacturer’s instructions. Gene expression analysis was performed using Affy Human U133Plus 2.0 at the Genomics and Proteomics Core Facility of the German Cancer Research Center (GPCF DKFZ, Heidelberg).
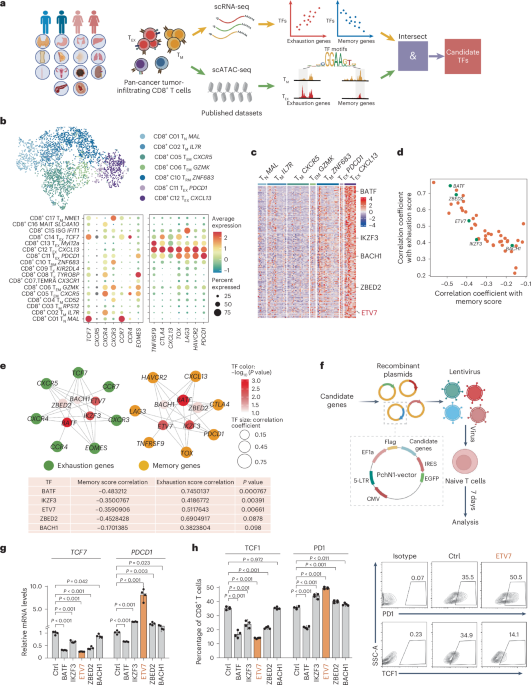
To test differences in lung metastases versus primary tumors, genes were ranked based on log fold change. GSEA was performed with the R package gage (v2.36.0) and default settings using the C2 curated gene sets from MSigDB (v7.4). For the top hit (Nagashima NRG1 signaling up) as well as our custom HER3 signature (Supplementary Table 3 and scRNA-seq analysis), GSEA was performed with the R package fgsea (v1.12.0) and default settings to produce the plots in Figs. 1d and 3e, respectively.
Western blottingCells were lysed in RIPA-based protein lysis buffer. Protein concentration was determined using a Pierce BCA Protein Assay kit (Thermo). After blocking for 1 h at room temperature with 1× Tris-buffered saline and Tween 20 buffer containing 5% BSA, membranes were incubated overnight with the appropriate primary antibodies diluted 1:1,000 in blocking buffer. Antibodies to phospho-HER3/ERBB3 (Tyr 1289; 21D3; rabbit monoclonal 4791), HER3/ERBB3 (D22C5; XP rabbit monoclonal 12708), FGFR1 (D8E4; XP rabbit monoclonal 9740), phospho-AKT (Ser 473; D9E; XP rabbit monoclonal 4060), AKT (pan; C67E7; rabbit monoclonal 4691), phospho-p44/42 MAPK (ERK1/ERK2; Thr 202/Tyr 204; D13.14.4E; XP rabbit monoclonal 4370), p44/42 MAPK (ERK1/ERK2; 137F5; rabbit monoclonal 4695), phospho-FAK (Tyr 397; D20B1; rabbit monoclonal 8556), FAK (rabbit polyclonal 3285) and GAPDH (14C10; rabbit monoclonal 2118) were purchased from Cell Signaling Technology. Monoclonal anti-α-tubulin T5168 was purchased from Sigma-Aldrich.
IHC analysisSections from fixed CDOs or tissues were obtained, processed and stained as previously described49, with minor modifications. The following antibodies were used: anti-EpCAM (Agilent Dako, clone Ber-EP4, 1:100), anti-human KRT19 (Agilent Dako, clone RCK108, 1:50), anti-human Ki-67 (Agilent Dako, clone Ki-67, 1:1,000), anti-ERα (Thermo Fisher Scientific, clone SP1, 1:50), anti-CDH1 (Agilent Dako, clone M3612, 1:30) and anti-vimentin (Agilent Dako, clone M7020, 1:1,000). For HER3 expression, we incubated samples with anti-HER3/ERBB3 (D22C5, XP rabbit monoclonal 12708, 1:50) at 4 °C overnight and used heat-induced antigen unmasking with damp heat at 90 °C with EDTA unmasking solution (pH 9 (1:10); 14747 Signal Stain) for 30 min. Sections were scanned using a Zeiss AxioScan, and representative images are shown.
Targeted next-generation sequencing of somatic mutationsGenomic DNA was extracted from CDOs and CDXs. DNA concentration was assessed by fluorimetric measurement using a QuBit 3.0, and the amount of amplifiable DNA (sequencing-grade quality) was determined using a quantitative assay (TaqMan RNaseP detection assay) on a StepOnePlus instrument (both Thermo Fisher Scientific). Samples were amplified using a custom-designed gene panel for BC50, covering the most recurrent mutations51. Library preparation and sequencing were performed using multiplex PCR-based Ion Torrent AmpliSeq (Thermo Fisher Scientific) and Ion S5XL technology, as previously described52.
Bulk RNA-seq of CDOsCTCs were collected, washed in PBS and lysed in RNA lysis buffer. RNA was extracted using a PicoPure RNA isolation kit following the manufacturer’s instructions. RNA concentration and quality were assessed by Bioanalyzer (Agilent). Libraries were prepared using 5 ng of total RNA with an NEBNext Single Cell/Low Input RNA Library Prep kit (New England Biolabs) following the manufacturer’s instructions. Library concentration was quantified with QuBit, and library size distribution was assessed by Bioanalyzer. Up to 15 libraries were pooled equimolarly and sequenced on a NovaSeq 6000 S1 (paired-end, 150 base pairs).
Bulk RNA-seq analysisBcl2fastq2 2.20 was used for conversion. Reads were trimmed for adapter sequences and aligned to the 1000 Genomes Phase 2 assembly of the Genome Reference Consortium human genome (build 37, version hs37d5) with STAR53 (v2.5.3a) using the following parameters: alignIntronMax: 500,000; alignMatesGapMax: 500,000; outSAMunmapped: within; outFilterMultimapNmax: 1; outFilterMismatchNmax: 3; outFilterMismatchNoverLmax: 0.3; sjdbOverhang: 50; chimSegmentMin: 15; chimScoreMin: 1; chimScoreJunctionNonGTAG: 0 and chimJunctionOverhangMin: 15. GENCODE gene annotation (GENCODE release 19) was used for building the index. BAM files were sorted using SAMtools54 (v1.6), and duplicates were marked with Sambamba55 (v0.6.5). Raw counts were generated using featureCounts56 (Subread version 1.5.3).
For calculation of normalized counts, mitochondrial RNA, tRNA, rRNA and all transcripts from the Y and X chromosomes were removed, and normalization was performed in analogy to transcripts per million.
Differential gene expression analysis was performed using DESeq2 (ref. 56; v1.26.0). The lfcshrink function was used to define differentially expressed genes (| log2 (fold change) |) of ≥1, adjusted P value of ≤0.05). The log2 (fold change) values (nonshrinked) were used for GSEA with clusterProfiler57 and the Molecular Signatures Database v7.411 as reference gene sets. Data handling was performed in R (v3.6.0) using RStudio (v1.4).
Gene expression analysis of human CTCs: scRNA-seqFrom cryopreserved vials of CTC samples, single live Lin–EpCAM+ cells were directly sorted by FACS into 100 µl of TRIzol (Thermo Fisher). Samples were immediately snap-frozen in liquid nitrogen and stored at −80 °C. For RNA isolation, TRIzol samples were thawed on ice and mixed with 20 µl of chloroform. After incubation at room temperature for 3 min, samples were centrifuged (12,000g, 5 min, room temperature) and immediately transferred on ice. The aqueous phase was collected and mixed with 0.4 µl of GlycoBlue (Thermo Fisher) in 75 µl of isopropanol, and samples were stored at −20 °C for at least 5 days. Samples were centrifuged (13,000g, 1 h, 4 °C), and the pellet was washed with 70% ethanol and centrifuged again (13,000g, 15 min, 4 °C). The pellet was resuspended in 5 µl of Smart-Seq2 buffer. Whole-transcriptome amplification was performed using the modified Smart-Seq2 protocol as previously described58. Libraries were constructed using a Nextera XT DNA Library Preparation kit (Illumina) according to the manufacturer’s instructions but using one-fourth of all volumes. Sequencing was performed on an Illumina HiSeq 2500 platform.
scRNA-seq analysisRaw data processing was performed with kallisto59 (v0.43.0). The kallisto index file was generated with a hg38 transcriptome fasta file (release-98) downloaded from Ensembl, and reads were then pseudoaligned to the transcriptome with kallisto in quant mode. The R package tximport60 (v1.14.2) was used to perform gene-level summaries, and the resulting count matrix was imported as a SingleCellExperiment object in R.
The R packages scater61 (v1.14.6) and scran62 (v1.14.6) were used to calculate quality control metrics and remove cells with less than 1 × 105 total counts, less than 2,500 detected features or a percentage of mitochondrial genes higher than 20%. Normalization and log transformation of the data were performed with the functions computeSumFactors and logNormCounts. Cells not expressing the epithelial marker EpCAM or expressing the leukocyte marker CD45 were removed, leaving 318 putative CTCs.
Cells were then separated into ERBB3hi and ERBB3lo populations based on the results of a k-means clustering (k = 2) on ERBB3 expression values (Fig. 3d). To define a HER3 signature, genes that were not expressed in at least 20% of cells were removed. Differentially expressed genes between the ERBB3hi and ERBB3lo populations were then computed using the pairwiseWilcox function from scran (FDR < 0.1). Resulting significant genes were intersected with genes whose expression showed a significant Pearson correlation (FDR < 0.1) with the expression of ERBB3 and protein-coding genes, yielding 592 upregulated genes and 6 downregulated genes (Supplementary Table 3). To see if the HER3 signature could be used to separate the ERBB3hi and ERBB3lo populations in the three individuals, z scores were computed for each signature gene for the two populations (\(z=\,\frac}}\)), and the mean over all genes was calculated. For better UMAP visualization and coloring of expression of different genes (Fig. 6a and Extended Data Figs. 6b,c and 10a), data were integrated with mutual nearest neighbors, as implemented in the fastMNN() function from the batchelor63 (v1.2.4) R package.
Genomic analysis of CDOs and participant-matched lesionsWhole-genome sequencing and whole-exome sequencing data were aligned and analyzed using the Sarek64 (v3.1.2) Nextflow65 (v22.10.7) workflow from the nf-core framework. Briefly, initial quality control and read trimming were performed with FASTQC (v0.11.9) and fastp66 (v0.23.2). Trimmed reads were then aligned to the GRCh38 reference genome using BWA-mem67 (v0.7.17-r1188). Aligned reads were further preprocessed using GATK4 (ref. 46; v4.3.0.0). Variants were called using Strelka2 (ref. 46; v2.9.10), Mutect2 or Manta68 (v1.6.0) using matching germline controls when possible. Variants were annotated using Ensembl VEP69 (v106.1). For subsequent analyses, only variants fulfilling the following criteria were selected: (1) called by both Strelka2 and Mutect2, (2) FILTER = = PASS, (3) at least 20 reads mapped in the germline control sample or in the tumor sample and (4) at least 2 reads mapped in the alternative allele in the tumor sample. Oncoplot was generated using R 4.3.0 and the maftools70 package (v2.16.0). A panel of most common and relevant mutated genes was defined based on previous studies on MBCPs25,30.
Gene expression analysis in longitudinal clinical lesionsRNA isolation from fresh-frozen or formalin-fixed paraffin-embedded tumor samples, quality control, stranded library preparation, sequencing, alignment of resulting reads and their summarization were all performed as previously described25. The obtained read counts for 22 longitudinally profiled tumor pairs were subjected to variance-stabilizing transformation using DESeq2 (ref. 71; v1.22.2), which was subsequently used as input for the PCA. Based on the PCA, the initial set was further reduced to 14 pairs, selecting samples that belong to the same cluster along the first PC axis.
Genome-wide CRISPRa screen to identify regulators of NRG1 dependencyThe human CRISPRa pooled library Set A (Addgene plasmid 92379; a gift from D. Root and J. Doench, Broad Institute of Harvard and MIT) was amplified as previously described72. CRISPR library complexity was assessed via next-generation sequencing using a HiSeq2000. For the validation experiments, individual gRNAs were cloned into pXPR_502 (Addgene 96923; a gift from J. Doench and D. Root) lentivector for CRISPRa experiments via restriction digest with BsmBI (New England Biolabs, R0739). CTC596 cells were transduced with lentiviral particles carrying dCas9-VP64 (lenti dCas9VP64_Blast, Addgene plasmid 61425; a gift from F. Zhang, Broad Institute of MIT and Harvard) at a multiplicity of infection of ∼0.6. After recovery, cells were selected with blasticidin (20 μg ml–1). CTC596 cells expressing dCas9-VP64 were transduced with the genome-wide Calabrese CRISPRa library in two independent experiments at a multiplicity of infection of ∼0.15. For each replicate, ∼200 million cells were transduced, achieving a representation of 300/500 cells per gRNA. After initial recovery for 2 days, cells were selected with puromycin (1 μg ml–1) for 4 days. For each replicate, cells were collected at t0 to determine baseline gRNA representation, t1 and t2. Genomic DNA was extracted using a Quick-DNA Midiprep Plus kit (Zymo Research, D4075) following the manufacturer’s instructions, and next-generation sequencing libraries were prepared as previously described72. Libraries were sequenced as a multiplexed pool on a HiSeq2000 (125 cycles read 1, 8 cycles index 1 (i7)). Data were analyzed using the PinAPL-Py web tool73. Briefly, after trimming of the adapters, reads were aligned to the reference library and counted. A gene score (SigmaFC) was then calculated by taking the sum of the log (fold change) values of all sgRNAs targeting that gene and multiplying it by the number of its sgRNAs that reached statistically significant enrichment. The Benjamini–Hochberg correction method was used for P value correction.
Statistics and reproducibilitySample sizes are similar to those reported in previous publications19,74,75, and no statistical method was used to predetermine sample size. Statistical analysis and data visualization were performed using GraphPad Prism (v10.3.0 and earlier) software, except for genomic and transcriptomic analysis and visualization, which were performed using R. Data collection and analysis were not performed blind to the conditions of the experiments. For in vivo treatment studies, mice were randomized before the start of treatment to ensure that each group started with an approximately equal mean tumor size. For comparison between two sample groups, statistical analysis was conducted using a two-tailed (paired or unpaired) Student’s t-test, two-tailed Mann–Whitney test and two-tailed Wilcoxon test, unless otherwise stated. For multiple comparisons, one- or two-way ANOVA was used. Data distribution was not formally tested. A P value of <0.05 was used as a cutoff for significance. Data are generally presented as mean ± s.e.m. of n = x experiments, with x indicating the number of independent experiments performed, which is noted in the figure legends.
Reporting summaryFurther information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
留言 (0)