Animals
All experiments were conducted in accordance with the National Institute of Health Guide for Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee of Boston Children’s Hospital and Harvard Medical School (protocol number IS00000270-6). All mice were kept under specific pathogen-free conditions within an environment controlled for temperature (20–22 °C) and humidity (40–70%), and were subjected to a 12 h light/dark cycle. The generation of Gabpa-dTAG knock-in mice was as described previously with some modifications23. Briefly, two-cell embryos (20 hpf) were injected with Gabpa donor DNA (30 ng μl−1), Cas9 mRNA (100 ng μl−1) and sgRNA (single guide RNA) (50 ng μl−1) using a Piezo impact-driven micromanipulator (Primer Tech). Then, two-cell embryos were incubated in KSOM (potassium simplex optimized medium) for 2 h before transfer into oviducts of pseudo-pregnant Institute of Cancer Research (ICR) strain mothers (Charles River). F0 chimera mice were backcrossed with wild-type C57BL/6J mice for at least two generations. Genotyping was performed using mouse tail lysed in lysis buffer (50 mM Tris–HCl, 0.5% Triton and 400 μg ml−1 Proteinase K) at 55 °C overnight. For F0 and F1 mice genotyping, the primers outside the homology arm are used. For genotyping of F2 and beyond, the inner primers are used. The primers are listed in Supplementary Table 6.
Droplet digital PCR (ddPCR) was used for detecting the copy number of Gabpa-dTAG knock-in alleles in F1 mice. Briefly, 250 ng purified DNA templates were digested by incubation with Haelll Enzyme (NEB) at 37 °C for 1 h and then inactivated at 80 °C for 5 min. A final 30 ng of DNA was used as the templates for PCR. Fkbp was used for knock-in detecting, and mRPP30 was used as a control. The primers are included in Supplementary Table 6. Only the mice with a single Fkbp copy (Supplementary Table 7) were used for further mating.
mES cell culture and establishment of Gabpa
dTAG/dTAG cell line
The laboratory-maintained ES-E14 cells45 were cultured on 0.1% gelatin-coated plates with 2i/LIF (MEKi/GSK3i/leukemia inhibitory factor) condition. Cells were grown in Dulbecco’s modified Eagle medium (Gibco, 11960069), supplemented with 15% foetal bovine serum (Sigma-Aldrich, F6178), 2 mM GlutaMAX (Gibco, 35050061), 1 mM sodium pyruvate (Gibco, 11360), 1× MEM NEAA (minimum essential medium nonessential amino acids) (Gibco, 11140050), 0.084 mM 2-mercaptoethanol (Gibco, 21985023), 1 mM sodium pyruvate (Gibco, 11360070), 100 U ml−1 penicillin–streptomycin (Gibco, 15140122), 1,000 IU ml−1 LIF (Millipore, ESG1107), 0.5 μM PD0325901 (Tocris, 4192) and 3 μM CHIR99021 (Tocris, 4423).
To establish GabpadTAG/dTAG cell line, Gabpa-HAL-FKBPF36V-2xHA-HAR and px330 were transfected into mouse (m)ES cells with Lipofectamine 2000 (Thermo, 11668030). Twenty-four hours later, cells were selected with puromycin (Gibco, A1113803) for another 48 h. Then, cells were cultured in puromycin-free medium for 1 week. Single clones were picked for genotyping and further analysis.
In vitro fertilization and embryo culture
Female mice (7–8 weeks) were superovulated through an initial injection of 7.5 IU pregnant mare serum gonadotropin (BioVendor, RP1782725000), followed 48 h later with a 7.5 IU injection of human chorionic gonadotropin (Sigma, C1063). Oocyte–cumulus complexes (OCCs) were collected 14 h post human chorionic gonadotropin injection. Sperm was collected from the cauda epididymis of adult male mice (8–12 weeks) 1 h before OCC collection. The sperm suspension was capacitated for 1 h in 200 μl human tubal fluid (HTF) medium (Millipore, MR-070-D). Subsequently, OCCs were exposed to spermatozoa for a 6 h incubation. The time when sperm was added to OCCs was considered as 0 hpf. Two-nuclear zygotes were cultured in the KSOM medium (Millipore, MR-106-D) under a humidified atmosphere of 5% CO2 at 37 °C for further development.
Western blot
A total of 1.5 × 106 cells were lysed in 100 µl RIPA lysis buffer and incubated on ice for 30 min. Ninety microlitres of supernatant was mixed with 12.5 µl 5× loading buffer, and heated at 98 °C for 15 min. Samples were run on NuPAGE 4–12% gel (Invitrogen, NP0322BOX) and transferred onto polyvinylidene fluoride transfer membrane. Primary antibodies used included anti-GABPA (1:4,000, Proteintech), anti-HA (1:1,000, CST) and anti-β-actin (1:5,000, CST, #4967). Secondary antibodies used included goat anti-Rabbit IgG (H + L) superclonal secondary antibody-HRP (Thermo Scientific, A27036, 1:2,000) and goat anti-mouse IgG (H + L) secondary antibody-HRP (Thermo Fisher Scientific, 31430, 1:2,000). Protein bands were detected with an enhanced chemiluminescence (ECL) kit (Thermo Fisher Scientific, 32209) and imaged by Tanon 4600SF Imaging System (Tanon).
Immunostaining and confocal microscope
Embryos were fixed with 4% paraformaldehyde/0.5% Triton for 20 min, followed by three times of washing with phosphate-buffered saline/0.1% Triton, then blocked in phosphate-buffered saline/1% bovine serum albumin/0.01% Triton for 1 h. Embryos were incubated overnight at 4 °C with primary antibodies: anti-GABPA (1:200, Proteintech, 21542-1-AP, lot no. 00018047), anti-HA (1:200, CST, 2367S), anti-GATA4 (1:200, R&D Systems, MAB2606-SP), anti-NANOG (1:200, Abcam, ab80892) and anti-CDX2 (1:500, R&D Systems, AF3665-SP). Secondary antibodies used included donkey anti-goat IgG (H + L) secondary antibody, Alexa Fluor 647 (Thermo Scientific, A-21447, 1:500), donkey anti-rabbit IgG (H + L) secondary antibody, Alexa Fluor 488 (Thermo Scientific, A-21206, 1:500) and Donkey anti-mouse IgG Secondary Antibody, Alexa Fluor 568 (Fisher Scientific, A10037, 1:500). After three times of washing, the embryos were incubated with secondary antibodies at room temperature (RT). DNA was stained with 10 μg ml−1 Hoechst 33342 (Sigma). The confocal microscope (Zeiss, LSM800) was used for fluorescence detection.
dTAG13 treatment
dTAG13 (Tocris, 6605) was reconstituted in DMSO to a 5 mM stock. For GabpadTAG/dTAG mES cell treatment, dTAG13 was diluted in mES cell culture medium to 0.5 μM. For GabpadTAG/dTAG embryo treatment, dTAG13 was diluted in KSOM to 1 μM. Embryos were washed with KSOM with dTAG13 at least three times, then cultured in KSOM with dTAG13 for further development.
CUT&RUN, ATAC and RNA-seq library preparation and sequencing
For mES cell CUT&RUN with more than 10,000 cells were resuspended in 50 μl washing buffer (20 mM HEPES pH 7.5, 150 mM NaCl, 0.5 mM spermidine and 1× protease inhibitor) with activated Concanavalin A magnetic beads (Polysciences, 86057-3) for 10 min at RT, then samples were incubated with anti-GABPA (1:40, Proteintech, 21542-1-AP, lot#00018047; note that this is the only lot that worked in CUT&RUN in our hands) overnight at 4 °C. For low-input mES cells and embryo CUT&RUN, some modifications were made. Briefly, mES cells, zona-free embryos or isolated ICM were resuspended in 50 μl washing buffer with activated Concanavalin A magnetic beads for 10 min at RT, then samples were fixed with 1% formaldehyde for 1 min. After three washes with washing buffer, samples were incubated with anti-GABPA overnight at 4 °C. Samples were incubated with 2.8 ng μl−1 pA-MNase (home-made) for 2 h at 4 °C. Subsequently, samples were incubated with 200 μl pre-cooled 0.5 μM CaCl2 for 20 min at 4 °C and quenched by adding 23 μl 10× stop buffer (1,700 mM NaCl, 20 mM EGTA, 100 mM EDTA, 0.02% digitonin, 250 µg ml−1 glycogen and 250 µg ml−1 RNase A). DNA fragments were released by incubation at 37 °C for 15 min. For both fixed and unfixed cells, 2.5 μl 10% SDS and 2.5 μl 20 mg ml−1 Proteinase K (Thermo Fisher) was added and incubated at 55 °C for at least 1 h for reverse crosslinking. DNA was extracted by phenol–chloroform followed by ethanol precipitation. The subsequent procedure was the same as described above. Sequencing libraries were prepared with NEBNext Ultra II DNA library preparation kit for Illumina (New England Biolabs, E7645S).
ATAC–seq was performed as previously described with some modifications46. Briefly, ES cells and isolated ICM were digested with adapter-loaded Tn5 for 15 min at 37 °C, and stopped by stop buffer (100 mM Tris pH 8.0, 100 mM NaCl, 40 µg ml−1 Proteinase K and 0.4% SDS) and incubated overnight at 55 °C. Five microlitres of 25% Tween-20 was added to quench SDS. Sequencing libraries were prepared with NEBNext High-Fidelity 2× PCR Master Mix (NEB, M0541S).
For RNA-seq, fresh ES cells or embryos were collected. Reverse transcription and complementary DNA amplification were performed with SMART-Seq v4 Ultra Low Input RNA Kit (Clontech, 634890), followed by cDNA fragmentation, adaptor ligation and amplification using Nextera XT DNA Sample Preparation Kit (Illumina, FC-131-1024).
All libraries were sequenced by NextSeq 550 system (Illumina) with paired-ended 75 bp reads (Supplementary Table 8).
Immunosurgery
ICMs were isolated as previously described47. Briefly, blastocysts at E3.5 or E4.5 stages were collected by removing the zona pellucida with Acidic Tyrode’s solution (Millipore). Embryos were then treated with anti-mouse serum antibody (Sigma-Aldrich, M5774-2ML, 1:5 dilution in KSOM) for 30 min at 37 °C. After washing three times with KSOM, embryos were treated with guinea pig complement (Millipore, 1:5 dilution in KSOM) for another 20 min at 37 °C. Then, the TE cells were removed by a glass pipette (the inner diameter is around 40–50 μm). E4.5 ICM refers to the mixture of EPI and PrE cells after removing TE cells with immunosurgery.
RNA-seq data analysis
The raw sequencing reads were trimmed with Trimmomatic48 (v0.39) to remove sequencing adaptors. Then, the reads were mapped to GRCm38 genome using STAR49 (v2.7.8a). Gene expression levels were quantified with RSEM50 (v1.3.1). To identify differentially expression genes, the DESeq251 (v1.32.0) package in R was used. The significantly differentially expressed genes were called with an adjusted P-value cut-off of 0.05, fold change cut-off of 2 and mean FPKM (fragments per kilobase of transcript per million mapped reads) cut-off of 1. GO enrichment was performed using R package clusterProfiler52. Gene Set Enrichment Analysis (GSEA) was performed using R clusterProfiler52 and enrichplot.
CUT&RUN data analysis
The raw reads were trimmed with Trimmomatic48 (v0.39) to remove sequencing adaptors, then mapped to GRCm38 reference genome using bowtie253 (v2.4.2). PCR duplicates were removed with Picard MarkDuplicates (v2.23.4). Reads with mapping quality less than 30 were removed. The mapped reads were further filtered to retain only proper paired reads with fragment length between 10 and 120. Peaks were called with MACS254 (v2.2.7.1). Reproducible peaks were generated with the irreproducible discovery rate (IDR) framework55 using two replicates, with IDR threshold of 0.05. For E4.5 ICM and ES cells, we further filtered the peaks to keep the ones with q value ≤10−30 to remove weak peaks. The signal tracks were generated with deeptools56 bamCoverage (v3.5.1) with bin size of 1 and normalized by CPM (counts per million). For z-score-normalized signal tracks, we first used bamCoverage with bin size of 100 to generate FPKM signals, then used a customized script to calculate the z score of each bin. For late two-cell and eight-cell GABPA ultralow-input CUT&RUN data, we noticed low mapping rates of the raw data. Further examination of the unmapped reads suggested they were environmental DNA from human, bacteria, vectors and so on, and due to the ultralow-input cells and small number of GABPA binding regions, the ratio of mapped reads from GABPA-bound DNA versus unmapped reads arising from environmental DNA was low. However, this would not affect the identification of GABPA peaks, since (1) the discarded reads were unmappable to mouse reference genome and (2) these peaks disappeared upon GABPA degradation.
The peaks were annotated with R package ChIPseeker57. Peaks within −1,000 to +500 around the TSS were considered as promoter peaks.
The heatmaps of binding profiles were calculated with deeptools56 computeMatrix (v3.5.1) using bigwig signal tracks as input and bin size of 10, and visualized in R with packages profileplyr and EnrichedHeatmap58.
ATAC–seq data analysis
ATAC–seq data were analysed with the ENCODE59 ATAC–seq pipeline with default parameters (v2.1.2, https://github.com/ENCODE-DCC/atac-seq-pipeline).
Motif enrichment analysis
Motif enrichment analysis was performed with HOMER60 (v4.11) findMotifsGenome.pl with mm10 reference and parameter size 200, using peaks file as input.
Motif occurrence analysis
Motif occurrence analysis was performed with HOMER60 (v4.11) annotatePeaks.pl with parameters ‘mm10 -size -2000,2000 -hist 20 -ghist’ for peak regions and parameters ‘mm10 -size -500,500 -hist 20 -ghist’ for regions around gene TSSs. The motif files were download from JASPAR database61 and manually converted to HOMER motif format. We used the log odds detection threshold of 6.0 for all TFs we analysed. The motif occurrence matrix was visualized in R with package EnrichedHeatmap58.
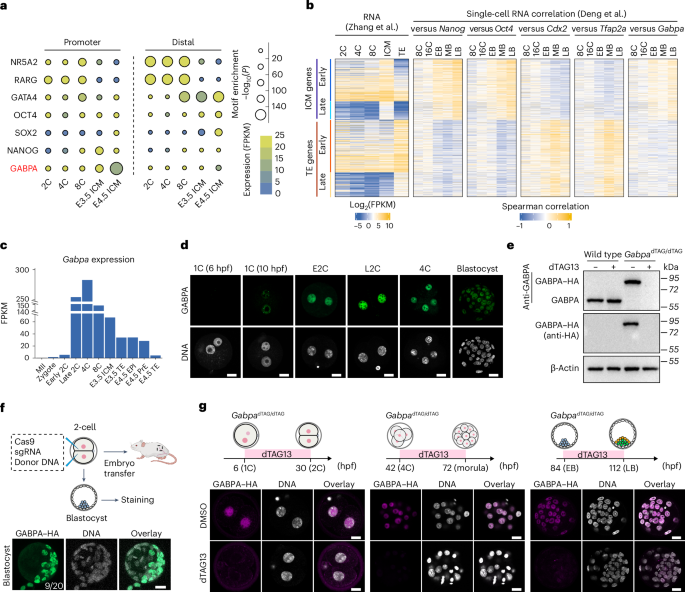
The JASPAR motif IDs for the TFs we analysed were as follows: GABPA, MA0062.2; OBOX, PH0121.1; DUX, MA0611.1; NFYA, MA0060.1; NR5A2, MA0505.1; OCT4, MA1115.1; SOX2, MA0143.1; NANOG, MA2339.1; TFAP2C, MA0524.2; GATA3, MA0037.4.
Identification of ICM/TE genes
ICM/TE genes were identified using bulk RNA-seq data of E3.5 ICM and E3.5 TE from Zhang et al.18. The DESeq251 (v1.32.0) package in R was used to find the DEGs between ICM and TE, with an adjusted P-value cut-off of 0.05 and fold change cut-off of 2. Genes with eight-cell FPKM ≥1 were defined as early ICM/TE genes, while genes with eight-cell FPKM <1 were defined as late ICM/TE genes.
Identification of EPI/PrE genes
EPI/PrE genes were identified using single-cell RNA-seq data of E4.5 embryos from Mohammed et al.62. The R package Seurat63 (v5.0.1) function FindMarkers was used to identify the DEGs between E4.5 EPI and E4.5 PrE, with an adjusted P-value cut-off of 0.05, fold change cut-off of 4 and cell expression percentage cut-off of 0.8. Genes with expression in at least 50% E3.5 single cells were considered as early EPI/PrE genes, while the others were considered as late EPI/PrE genes.
Statistics and reproducibility
Student’s t-tests for graph analysis were performed with Microsoft Excel (2016). Individual data points are shown as dots in the figure panels involving the Student’s t-test. Data distribution was assumed to be normal, but this was not formally tested. For the immunofluorescence and western blot experiments, at least three independent repetitions were performed with consistent results, and representative data were presented. P values < 0.05 were considered statistically significant. No statistical methods were used to pre-determine sample sizes, but our sample sizes are similar to or greater than those reported in previous publications8. No data were excluded from the analyses. The experiments were not randomized. Data collection and analysis were not performed blind to the conditions of the experiments.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.


留言 (0)