
記住我
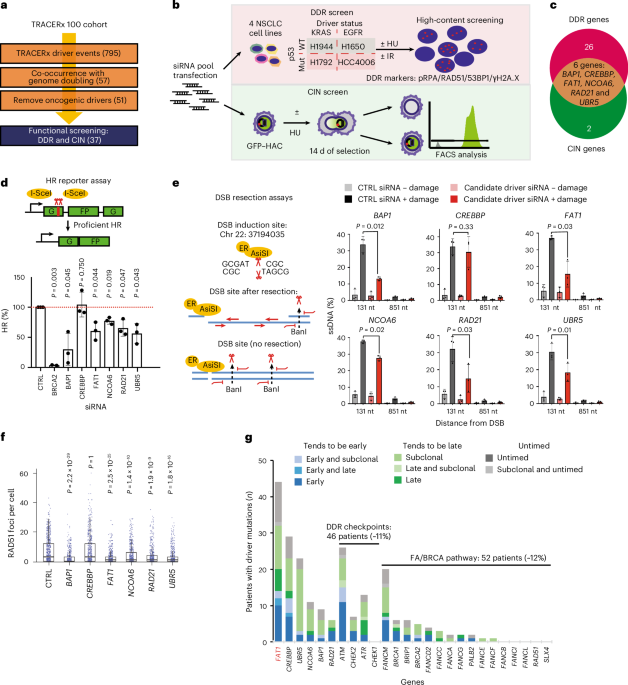

To identify alterations that correlate with WGD and CIN in NSCLC, we analysed multiregional WES data from tumours obtained from the first 100 patients within the lung TRACERx study3. Here, we identified 795 driver events in 91 genes, excluded known oncogenes that could not be modelled appropriately by genetic depletion approaches and focused on 37 tumour suppressor gene mutations that co-occurred with WGD (Fig. 1a). Pathway analysis revealed cellular processes related to genome maintenance, such as the DDR, transcription and chromatin remodelling (Supplementary Fig. 1)17,18,19. To assess how these genes contribute to genome integrity, we performed a multi-parametric RNA interference (RNAi) screen using high-content imaging in four different lung cancer cell lines harbouring mutations in KRAS, EGFR and TP53 to reflect the mutational landscape of the TRACERx cohort (Fig. 1b (top) and Extended Data Fig. 1a). DNA double-strand breaks (DSBs) induced by ionizing radiation, as well as replication stress induced by hydroxyurea, were chosen to model genotoxic stress. In parallel, the impact on chromosome loss was investigated utilizing a human artificial chromosome (HAC) reporter system20 (Fig. 1b (bottom) and Extended Data Fig. 1b). These combined approaches enabled the identification of six tumour suppressor driver genes—namely BAP1, CREBBP, FAT1, NCOA6, RAD21 and UBR5—as regulators of the DDR and maintenance of chromosomal stability in NSCLC (Fig. 1c). In addition, we also confirmed previously reported roles of WRN, FANCM, DICER1, SMARCA4/BRG1, ARID1B, ARID2, KDM5C and ATRX in the DDR21,22,23,24,25,26 (Extended Data Fig. 1a).
Fig. 1: DDR and CIN loss-of-function screen of genome doubling-associated drivers from the TRACERx 100 cohort.
a, Flow chart depicting candidate gene selection for the DDR and CIN screens. b, Schematic of the design of the DDR and CIN screens. c, Venn diagram showing the six driver genes contributing to DDR and CIN. d, Validation of the six candidate genes by DR-GFP homologous recombination reporter assay; BRCA2 serves as a positive control. HR efficiencies are normalized to those of control samples. Statistical significance was determined by two-sided, one-sample t-test. The data represent means ± s.d. (n = 3 biological replicates, except for BRCA2, for which n = 2). e, Validation of the six candidate genes by DIvA U2OS-AsiSI site-directed resection assay. Statistical significance was determined by two-sided paired t-test. The data represent means ± s.d. (n = 3 biological replicates). f, Box plots quantifying RAD51 foci formation in A549 cells following depletion of the six candidate genes, following 6 Gy ionizing irradiation and 1 h of recovery. The box edges represent interquartile ranges, the horizontal lines represent median values and the ranges of the whiskers denote 1.5× the interquartile range (n = 3 biological replicates; >150 cells quantified per biological replicate). Statistical significance was determined by Kruskal–Wallis test with Dunn’s multiple comparison test. g, Driver mutation distribution and mutational timing of the six candidate genes in the TRACERx 421 cohort. ATM, CHEK2, ATR, CHEK1 and members of the Fanconi anaemia (FA)/BRCA pathway are included for comparison. FAT1 is highlighted in red. CTRL, control; EGFR, epidermal growth factor receptor; FACS, fluorescence-activated cell sorting; HU, hydroxyurea; IR, ionizing radiation; mut, mutant; nt, nucleotides; WT, wild type.
The effect of these genes on the DDR was initially validated using two orthogonal approaches; the DR-GFP HR reporter assay27 and a site-specific DSB-generating endonuclease system (DIvA)28. Loss of BAP1, FAT1, NCOA6, RAD21 or UBR5, but not CREBBP, was associated with HR repair deficiency and impaired single-stranded DNA (ssDNA) resection—a step required for accurate HR repair (Fig. 1d,e and Supplementary Fig. 2). Next, we assessed early HR repair signalling 1 h after 6 Gy ionizing radiation in both A549 and H1944 cells. A marked decrease in the formation of RAD51 ionizing radiation-induced foci (IRIFs) was observed following depletion of BAP1, FAT1, NCOA6, RAD21 or UBR5 (Fig. 1f, Extended Data Fig. 2a,b and Supplementary Figs. 3 and 4). These alterations in HR efficiency could not be fully explained by cell cycle changes (Extended Data Fig. 2c), which dictate the selection of activating HR or non-homologous end-joining (NHEJ) repair, suggesting that these genes may be involved in HR directly. FAT1 was prioritized as a top candidate of clinical relevance, as inactivating mutations in FAT1 were highly recurrent (~10%) in the TRACERx 421 cohort (comprising 421 patients and 1,644 tumour regions), with a notable proportion of FAT1 mutations occurring early before WGD (Fig. 1g and Supplementary Fig. 5a–c). Notably, ~20% of the lung TRACERx cohort were found to harbour other inactivating mutations that impair HR efficiency (Fig. 1g and Extended Data Fig. 2d).
FAT1 alterations are positively selected in lung cancerTo quantify whether mutations in FAT1 were under positive selection, we measured the enrichment of FAT1 mutations before and after WGD using the ratio of the observed number of non-synonymous substitutions per non-synonymous site to the number of synonymous substitutions per synonymous site (dN/dS)29. Estimates of dN/dS above or below 1 suggest positive or negative selection, respectively, whereas estimates overlapping 1 imply that there is no evidence of selection. FAT1 mutations, which occurred more frequently in lung squamous cell carcinoma (LUSC) compared with lung adenocarcinoma (LUAD), were under greater positive selection before WGD occurrence in LUSC (Fig. 2a and Extended Data Fig. 3a). In the TRACERx 421 cohort, an enrichment of copy number deletion events was identified around the FAT1 genomic locus 4q35.2 only in patients with clonal WGD, indicating positive selection of 4q35.2 loss (Fig. 2b and Extended Data Fig. 3b–d). In LUSC, FAT1 promoter hypermethylation events reducing FAT1 expression levels and FAT1 copy number loss events were observed in the same tumours (Extended Data Fig. 3e–g). Furthermore, we detected a significant occurrence of mirrored subclonal allelic imbalance (MSAI) at the 4q35.2 locus, suggesting parallel evolution (Fig. 2c). Among genes encoded at the 4q35.2 locus, FAT1 has the lowest Genome Aggregation Database loss-of-function score in germline samples, implying that FAT1 loss is the least tolerated event within 4q35.2 (Fig. 2d). These results highlight the importance of FAT1 alterations.
Fig. 2: FAT1 loss of function is enriched in the TRACERx 421 cohort and leads to an elevated mitotic error rate and WGD.
a, Top, schematic of dN/dS ratio analysis. Bottom, results of dN/dS ratio analysis in the TRACERx 421 cohort, demonstrating that FAT1 truncation mutations are selected early in LUSC tumour evolution. The data points represent estimated dN/dS ratios and the error bars represent 95% confidence intervals calculated using the genesetdnds function from the R package dNdScv. The TRACERx 421 cohort comprised 233 males and 188 females (421 patients total), corresponding to a 55:45 male:female ratio. 93% of the cohort was from a White ethnic background and the mean age of the patients was 69 years, ranging between 34 and 92 years. Written informed consent was obtained. None of the patients was compensated for their involvement in the study. b, Genomic identification of significant targets In cancer (GISTIC) analysis of LUAD (141 patients) and LUSC tumours (80 patients) in TRACERx with clonal WGD only, demonstrating that SCNA loss at the FAT1 genomic locus (4q35.2; red text and highlighted) is positively selected in tumours with clonal WGD only. SCNA loci overlapping with common or rare chromosome fragile sites72,73 are annotated (in blue for common fragile sites and in green for rare fragile sites). c, MSAI analysis illustrating that the genomic region of chromosome 4 that harbours the FAT1 gene (arrows) is frequently lost in LUSC. Statistical significance was determined by Fisher’s exact test. In the schematic at the top, paternal and maternal chromosomes are indicated in blue and red, respectively. d, Top, schematic illustrating the location of the FAT1 gene on chromosome 4, together with other 4q35.2 genes within the frequently lost 4q35.2 genomic region. Bottom, selection pressures against losing genes. The data are from the Genome Aggregation Database (gnomAD) and demonstrate high selective pressure against deletion of the FAT1 genomic locus within 4q35.2. exp, expected; LOF, loss of function; obs, observed.
FAT1 ablation reduces HR efficiencyConsidering the frequency of FAT1 alterations in NSCLC (Figs. 1g and 2, Extended Data Fig. 3 and Supplementary Fig. 5) and its potential role in genome maintenance (Fig. 1d–f), we further elucidated at which stage FAT1 acts in the DSB repair pathway by systematically investigating which mediators were affected by FAT1 depletion 1 h post-ionizing radiation. FAT1 knockdown did not impact IRIFs of early DSB repair mediators, including phosphorylated ATM, γH2A.X and 53BP1 oligomerization, which are associated with NHEJ22 (Fig. 3a and Supplementary Fig. 6a,b). However, IRIF formation of CtBP interacting protein (CtIP), which is responsible for initiating ssDNA resection22, was significantly impaired by FAT1 knockdown (Fig. 3a and Supplementary Fig. 6c). FAT1 depletion also reduced IRIF formation of the key HR mediator breast cancer type 1 susceptibility protein (BRCA1) in G2/M cells, using centromere protein F-positive staining as a marker (Fig. 3a and Supplementary Fig. 6d). CRISPR knockout of FAT1 in H1944 or A549 cells impaired RAD51 IRIF formation, but not γH2A.X (Extended Data Fig. 4a,b and Supplementary Fig. 6e). A time-course post-ionizing radiation demonstrated that FAT1 depletion was associated with persistent DNA damage, as manifested by the increased frequency of 53BP1 nuclear bodies (Extended Data Fig. 4c).
Fig. 3: FAT1 loss attenuates HR repair.
a, Box plots demonstrating the impact of FAT1 siRNA knockdown on early DNA damage signalling and 53BP1 binding in A549 cells. The boxes represent interquartile ranges, the black and red bars represent median and mean values, respectively, and the ranges of the whiskers denote 1.5× the interquartile range. Statistical significance was determined by two-sided Wilcoxon rank-sum test (n = 3 biological replicates). The total numbers of cells quantified per condition were as follows: n ≥ 370 (pATM), n ≥ 438 (γH2A.X), n ≥ 448 (53PB1), n ≥ 560 (CtIP) and n ≥ 218 (BRCA1). b, Schematic of the FAT1 functional domains. The full-length FAT1 protein is 4,588 amino acids. c, RAD51 IRIF formation following 6 Gy ionizing radiation and 1 h of recovery in FAT1 CRISPR knockout (sgFAT1) versus control A549 cells with overexpression of HA–FAT1ICD versus pcDNA3.1. The boxes represent interquartile ranges, the black and red bars represent median and mean values, respectively, and the ranges of the whiskers denote 1.5× the interquartile range. Statistical significance was determined by two-sided Kruskal–Wallis test followed by Dunn’s test with Bonferroni correction (n = 3 biological replicates). d–f, Top, cartoons depicting examples of HRD-related large-scale transition (LST; d), telomeric allelic imbalance (TAI; e) and LOH (f). Bottom left, Permutation analysis showing a correlation between FAT1 CNA and HRD-related genomic signatures based on TCGA LUAD data. Red lines indicates 90 and 95% confidence intervals, blue line indicates observed correlation value. Bottom right, FAT1 driver mutation scores for these respective genetic alterations, based on TRACERx LUAD data. For the TRACERx LUAD data, tumour numbers were as follows: n = 212 (WT) and n = 17 (mut). In the box and whisker plots, the boxes represent interquartile ranges, the lines represent median values and the ranges of the whiskers denote 1.5× the interquartile range. Statistical significance was determined by two-sided mixed-effects linear model with purity as a fixed covariate and tumour ID as a random variable. g, Top, cartoon showing the design of the EJ5–GFP distal end-joining reporter integrated in U2OS cells. Bottom, 53BP1 siRNA knockdown, but not FAT1 knockdown, affects the distal end-joining rate. The data represent means ± s.d. Statistical significance was determined by two-sided repeated measures one-way analysis of variance (ANOVA) with Holm–Šidák correction (n = 5 biological repeats). h, Top, cartoon showing the design of the EJ2–GFP alternative end-joining reporter integrated into U2OS cells. Bottom, FAT1 siRNA knockdown significantly reduces the alternative end-joining efficiency. The data represent means ± s.d. Statistical significance was determined by two-sided paired t-test (n = 4 biological repeats). EGF, epidermal growth factor-like domain; LAMG, laminin G-like domain; NLS, nuclear localization signal.
Although the size of full-length FAT1 (4,588 amino acids) limits its ectopic expression, the reduction in RAD51 foci formation could be partially rescued by overexpression of the FAT1 carboxy (C)-terminal intracellular domain30 (HA–FAT1ICD; amino acids 4,202–4,588), which exhibited nuclear localization, suggesting that nuclear FAT1 may promote efficient HR (Fig. 3b,c and Extended Data Fig. 4d,e). FAT1-knockout A549 cells were more sensitive to genotoxic stress induced by poly(ADP-ribose) polymerase inhibitors, cisplatin and hydroxyurea (Extended Data Fig. 5a,b). By analysing both the TRACERx and The Cancer Genome Atlas (TCGA) LUAD datasets, we observed a correlation between FAT1 loss and HR deficiency (HRD)-related genomic signatures, including elevated telomeric allelic imbalance (TAI)31, large-scale transitions (LST)32 and loss of heterozygosity (LOH)33 (Fig. 3d–f). Using established reporters for distal and alternative end-joining activities, respectively34, we confirmed that transient FAT1 siRNA depletion reduced the alternative end-joining efficiency without significantly reducing distal end joining (Fig. 3g,h). Utilizing WGS data from Genomics England, no significant difference was observed in ID6 and SBS3 mutational profiles (Extended Data Fig. 5c,d and Supplementary Fig. 7a,b), both of which are mutation signatures associated with NHEJ activity35.
FAT1 ablation leads to structural and numerical CINIndeed, FAT1 depletion resulted in an increased fork collapse rate and HAC loss (Fig. 4a and Extended Data Fig. 1b). Our analysis of the TCGA and Genomics England datasets revealed that FAT1 loss correlated with an increased weighted genome instability index score and total mutational burden, indicating elevated structural and numerical CIN (Fig. 4b,c). To validate this observation, we used U2OS and type 2 pneumocyte (T2P) cells to investigate the formation of micronuclei and 53BP1 nuclear bodies in G1 daughter cells, both established markers of unresolved replication stress and HRD36,37,38,39. FAT1 ablation at baseline and under replication stress induced by either low-dose aphidicolin or a short pulse of hydroxyurea exacerbated the formation of cyclin A-negative (G1-specific) 53BP1 nuclear bodies and micronuclei (Fig. 4d–h and Extended Data Fig. 6a). Notably, acentric micronucleus formation was significantly elevated following FAT1 depletion in A549 and U2OS cells following replication stress (Fig. 4f,g). Under these conditions, FAT1 ablation also resulted in an increased mitotic error rate at baseline and under replication stress, which manifested as an increased formation rate of chromatin bridges and lagging chromosomes (Fig. 5a and Extended Data Fig. 6b). Utilizing the DIvA site-directed DSB system28,40, we further demonstrated that FAT1 depletion resulted in increased illegitimate translocation of two DSBs induced on chromosome 17 (Fig. 5b). Concurrently, higher rates of structural chromosomal aberrations, including radial chromosomes and chromatid gaps, were observed upon FAT1 loss (Fig. 5c and Extended Data Fig. 6c). FAT1 silencing also reduced mitotic fidelity, as evidenced by deviations in the modal chromosome number (Fig. 5d–f and Extended Data Fig. 6d).
Fig. 4: FAT1 loss elevates replication stress and micronuclei.
a, FAT1 knockout exacerbates replication fork stalling in A549 cells. Top, scheme of the nucleotide labeling used to measure replication fork stalling. Bottom (left) quantification; (right), representative image for the DNA fibre experiments. The data represent means ± s.d. Statistical significance was determined by two-sided paired t-test (n = 3 biological replicates; >600 forks counted in total). Scale bars, 20 µm. b, TCGA LUAD analysis showing that FAT1 copy number loss is significantly correlated with weighted genome instability index measurements. The blue lines indicate FAT1 loss and the red dotted lines indicate the 90 and 95% confidence intervals. Confidence intervals were generated using computational permutation analyses. c, Box plot comparing the numbers of indels in FAT1 WT versus mutated tumours in the Genomics England LUAD and LUSC cohorts. The boxes represent interquartile ranges, the lines represent median values and the ranges of the whiskers denote 1.5× the interquartile range. Statistical significance was determined by two-sided Wilcoxon rank-sum test. n = 818 (WT) and n = 16 (mut). d, Transient FAT1 siRNA knockdown induces the formation of 53BP1 bodies in cyclin A-negative U2OS cells following 4 mM hydroxyurea for 5 h and recovery for 24 h. Statistical significance was determined by two-sided Wilcoxon rank-sum test. Scale bars, 10 μm. The red bars in the graph to the left represent mean values (n = 5 biological replicates). e,f, Transient FAT1 siRNA knockdown in U2OS cells induces the formation of total micronuclei with or without replication stress induced by 5 h of 4 mM hydroxyurea followed by 24 h recovery (e), as well as the formation of both acentric and centromeric micronuclei following the hydroxyurea treatment (f). The data represent means ± s.d. Statistical significance was determined by one-way ANOVA with Bonferroni correction. Biological repeats: n = 8 (e) and n = 4 (f). g,h, FAT1 loss elevates the rate of micronuclei formation in response to replication stress induced by 0.2 µM aphidicolin treatment (24 h) following FAT1 CRISPR knockout in A549 cells (g) or transient siRNA knockdown in T2P cells (h). The data represent means ± s.d. Statistical significance was determined by two-sided Student’s t-test. Biological repeats: n = 4 (total micronuclei in g), n = 3 (centromeric and acentric micronuclei in g) and n = 8 (h).
Fig. 5: FAT1 loss increases structural CIN and chromosome numbers.
a, Transient FAT1 knockdown significantly increases the mitotic error rate (lagging chromosomes plus DAPI bridges; left; data represent means ± s.d.) and the occurrence of nucleoplasmid bridges (middle; red bars represent mean values) in U2OS cells after 5 h treatment with 4 mM hydroxyurea and 24 h recovery. Statistical significance was determined by one-way ANOVA with Bonferroni correction (left) or Dunn’s test (middle). Right, selected maximum projection images following FAT1 knockdown, showing DAPI-stained mitotic U2OS cells following treatment with 4 mM hydroxyurea and 24 h recovery. Scale bars, 5 μm. Over 100 mitotic cells were scored across three biological replicates. b, Representative PCR-based semi-quantitative DIvA U2OS-AsiSI translocation assay. Transient FAT1 siRNA knockdown increases illegitimate repair products. PCR products generated from the uncut region and the legitimate repair product were used as the loading control. n = 3 biological replicates. c, Histogram (left) and representative images (right) showing that A549 cells with FAT1 loss exhibit a significantly increased number of chromosomal aberrations upon challenge with replication stress induced by 0.2 µM aphidicolin (APH) treatment. Scale bar, 5 μm. The data represent means ± s.d. Statistical significance was determined by one-way ANOVA with Holm–Šidák correction. A total of 60 metaphases were scored across three biological replicates per condition. Blue and red arrows indicate radial chromosomes and chromatid gaps, respectively. d–f, Transient FAT1 siRNA knockdown causes a significant numerical deviation in chromosome number in H1944 cells, as determined by multiple methodologies, including clonal fluorescence in situ hybridization (d), ImageStream high-throughput flow cytometry (e) and metaphase spreads (f). The histogram data represent means ± s.d. For the box plots, the boxes represent interquartile ranges, the black and red lines represent median and mean values, respectively, and the ranges of the whiskers denote 1.5× the interquartile range. Statistical significance was determined by two-sided Wilcoxon rank-sum test (d) or two-sided paired t-test (e). n = 3 biological replicates for all cases. bp, base pairs. NT, non-targeting.
Since FAT1 mutations are both common in lung cancer and evolutionarily selected before WGD (Fig. 2a–c), we experimentally validated the mitotic defect associated with FAT1 loss using the near-triploid (3N) LUAD cell line PC9 (which harbours an in-frame deletion at exon 19 of the epidermal growth factor receptor-encoding gene) and its isogenic WGD hexaploid (6N) clone12. Transient FAT1 depletion in PC9 cells significantly elevated the rate of stalled replication forks and mitotic errors, further confirming the involvement of FAT1 in genome maintenance (Extended Data Fig. 6e–g). Despite resulting in an elevated replication fork collapse rate, reduced formation of interphase and mitotic Fanconi anaemia complementation group D2 (FancD2) foci was also observed in FAT1-depleted WGD PC9 cells, suggesting a failure to recover from replication stress, leading to structural CIN (Extended Data Fig. 6e,g–j).
FAT1 depletion leads to mitotic errors and WGDWGD events were highly prevalent in the lung TRACERx 421 cohort (84% of LUSC and 77% of LUAD) and FAT1 driver mutations were selected before WGD in LUSC tumours (Fig. 2a). However, FAT1 mutations and WGD did not significantly co-occur in the TRACERx 421 cohort (Fisher’s exact test; P = 0.179) (Extended Data Fig. 7a). This was probably due to the presence of other HR-related gene alterations (~20% of patients; Fig. 1g and Extended Data Fig. 2d), which also can contribute to WGD. To investigate whether FAT1 alterations drive WGD, we used the PC9 lung cancer model to quantify the proportion of actively replicating cells with >6N genome content (the basal ploidy of PC9 is 3N). Increased 5-ethynyl-2′-deoxyuridine (EdU) incorporation rates (Fig. 6a,b), as well as a significant increase in loading of the replicative helicase MCM7 beyond 6N, were observed in FAT1-knockout PC9 cells (Extended Data Fig. 7b), both indicating a second replication event post-6N and suggesting that FAT1 loss is associated with WGD. These observations were independent of p53 mutational status, as similar results were obtained in TP53 wild-type, near diploid, untransformed retinal pigment epithelial-1 (RPE-1) cells immortalized with the human telomerase reverse transcriptase subunit (hTERT) (Extended Data Fig. 7c).
To identify the cause of WGD following FAT1 depletion, we monitored cells using live-cell microscopy, tracking at single-cell resolution. Reported causes of WGD include cytokinesis defects, endoreplication, mitotic bypass and cyclin B1 dysregulation during G2 (refs. 41,42,43,44). No change in cyclin B1 level was observed upon FAT1 knockdown in G2, ruling out cyclin B1 dysregulation as the cause of WGD in the absence of FAT1 (Extended Data Fig. 7d,e). DNA synthesis, measured by EdU incorporation, was also unaltered in control versus FAT1-knockout WGD cells transiently blocked in mitosis using nocodazole (Extended Data Fig. 7f), thereby ruling out a role for FAT1 loss in driving WGD through endoreplication in a manner similar to that of cyclin E amplification reported recently44. To investigate mitotic bypass, we used the hTERT RPE-1 cell line expressing both H2B-mTurquoise and fluorescent, ubiquitination-based cell cycle indicator (FUCCI) (hereafter FUCCI–RPE-1 cells) for live-cell imaging (Supplementary Video 1). FAT1 depletion, irrespective of the induction of aphidicolin-induced replication stress, did not cause a significant increase in the rate of mitotic bypass (Fig. 6c and Supplementary Fig. 8). In contrast, FAT1-depleted cells demonstrated an elevated rate of cytokinesis failure, suggesting defects in this final step of cell division as the cause of the WGD associated with FAT1 deficiency (Fig. 6d, Extended Data Fig. 7g and Supplementary Video 2). FAT1 depletion was also associated with an increased rate of nuclear shape abnormalities in daughter cells after normal mitoses (Fig. 6e and Supplementary Video 3). Similarly, after FAT1 depletion, increases in multinucleation and nuclear morphology alterations were observed in fixed U2OS and RPE-1 cells (Extended Data Fig. 7h,i) and WGD PC9 cells, respectively, after replication stress exposure (Extended Data Fig. 8a).
Fig. 6: FAT1 loss leads to an elevated mitotic error rate and results in WGD.
a, Representative dot plots demonstrating FAT1 ablation in PC9 cells and assessment of EdU incorporation beyond the normal G2 phase, to visualize WGD. b, Top, representative western blot validating FAT1 knockout. Bottom, quantification of EdU incorporation beyond the normal G2 population showing that FAT1 knockout significantly increases the WGD population in PC9 cells. The data represent means ± s.d. Statistical significance was determined by one-way ANOVA with Bonferroni correction. Biological repeats: n = 7 (sgNT), n = 5 (sgFAT1 clone 1) and n = 4 (sgFAT1 clone 2). c, Schematic (left) and histogram (right) showing the impact of transient FAT1 knockdown in TERT RPE-1 cells on the promotion of WGD through mitotic bypass, as determined by live-cell imaging. The data represent means ± s.e.m. Statistical significance was determined by one-way ANOVA with Bonferroni correction. Biological repeats: n = 3 (with aphidicolin treatment) and n = 6 (without aphidicolin treatment). d,e, Schematics (left) and histograms (right) showing that transient FAT1 siRNA knockdown in TERT RPE-1 cells increases the rates of cytokinesis failure (d) and nuclear shape deformation (e), as determined by 30× magnification live-cell microscopy imaging at 20 min intervals. The data represent means ± s.e.m. Statistical significance was determined by two-sided paired t-test, At least 200 mitotic events were tracked per condition over five biological replicates. YFP, yellow fluorescent protein.
To delineate whether structural CIN precedes nuclear shape abnormalities, we performed live-cell spinning-disk confocal microscopy on FUCCI–RPE-1 cells in G2, allowing us to determine the timing and outcome of the pending mitosis and the fate of respective daughter cells at high resolution with low phototoxicity. We observed that FAT1 depletion not only increases chromatin bridge formation rates (Extended Data Fig. 8b, left) but reduces the maintenance of normal nuclear morphology following mitotic chromosomal bridge formation (Extended Data Fig. 8b (right) and Supplementary Videos 4 and 5). Next, we investigated the long-term outcome of daughter cells with deformed nuclear morphology, by observing the heritability and recurrence of nuclear deformities and mitotic errors in respective daughter cells following mitosis (Extended Data Fig.
留言 (0)