
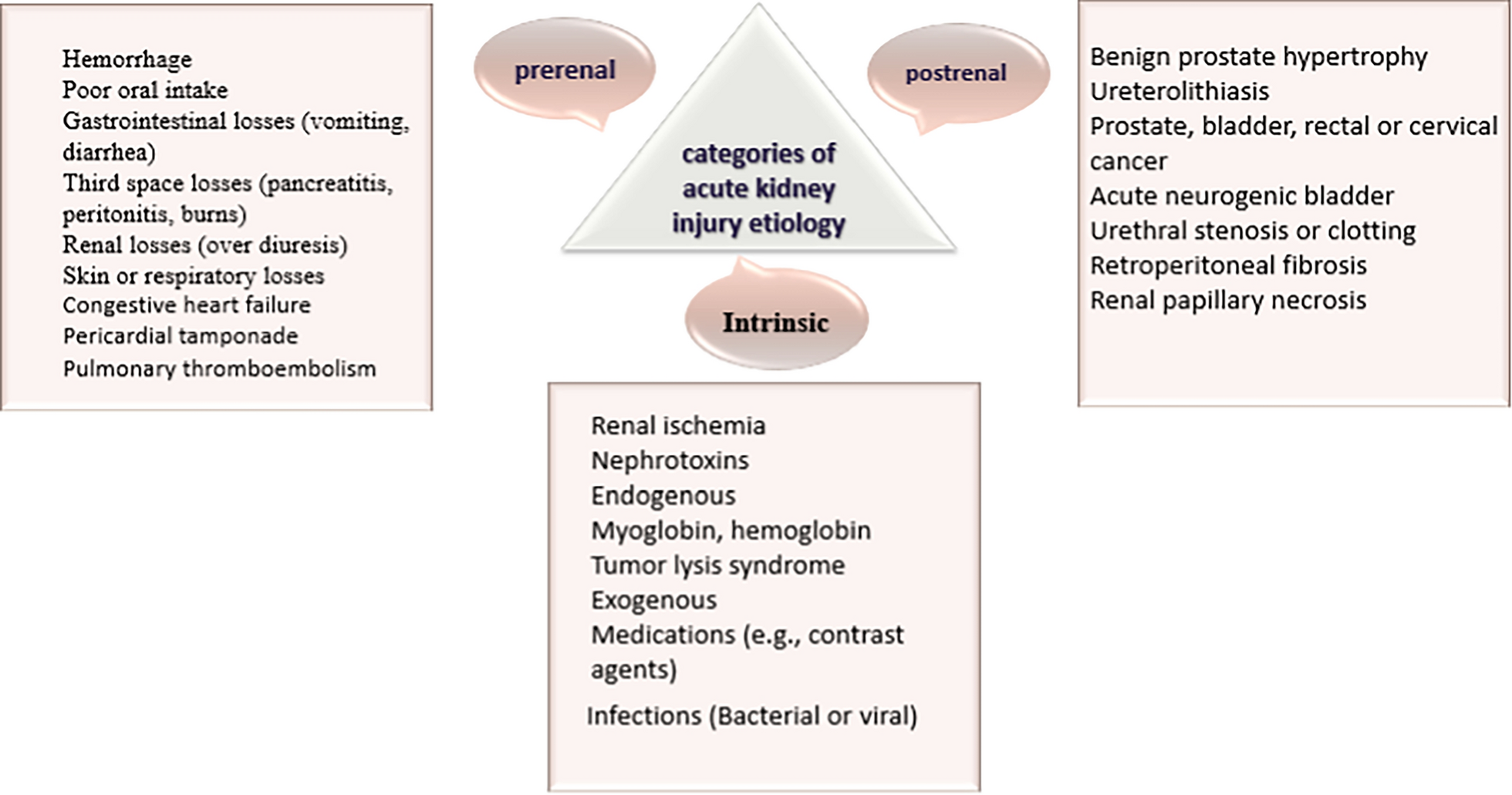
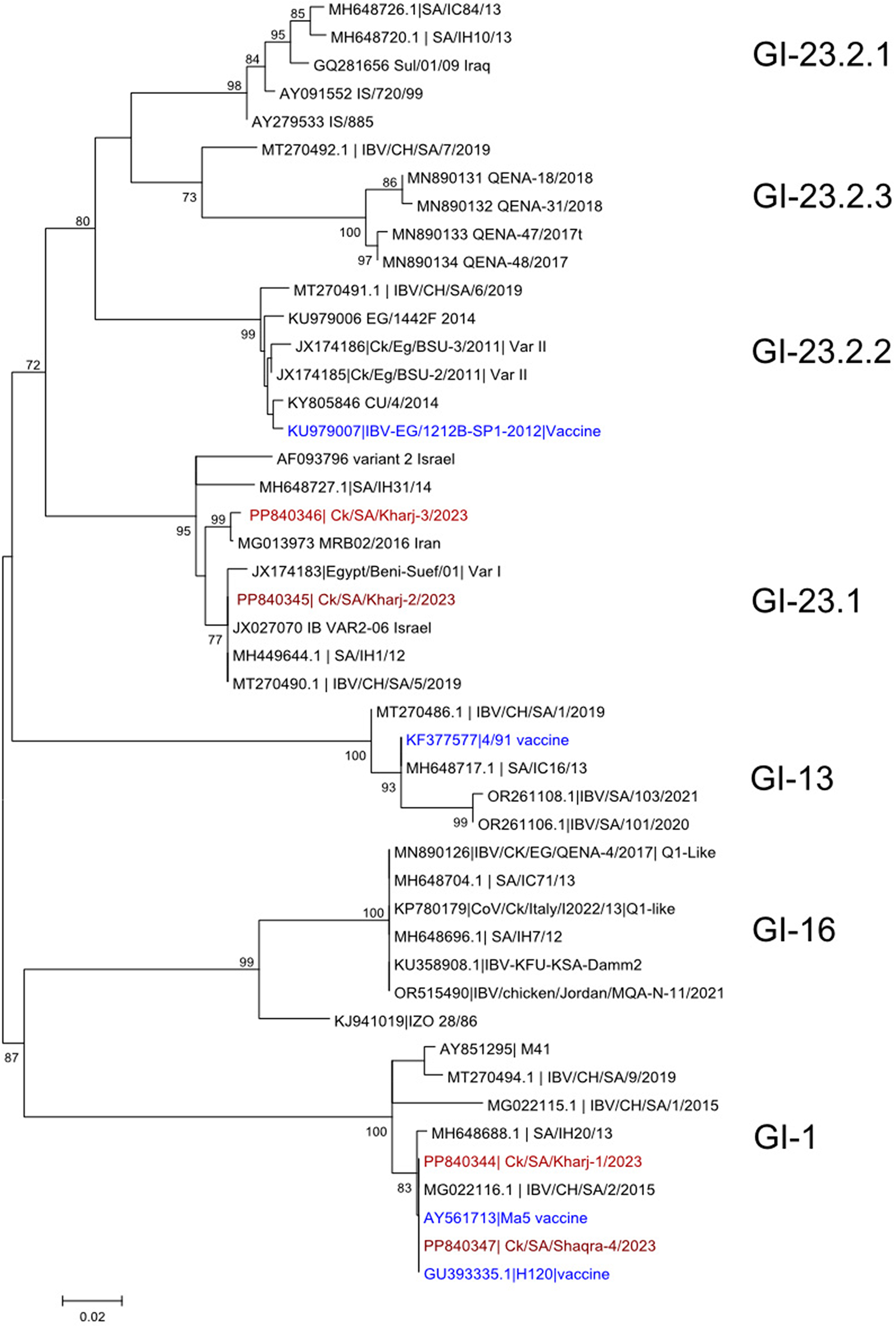
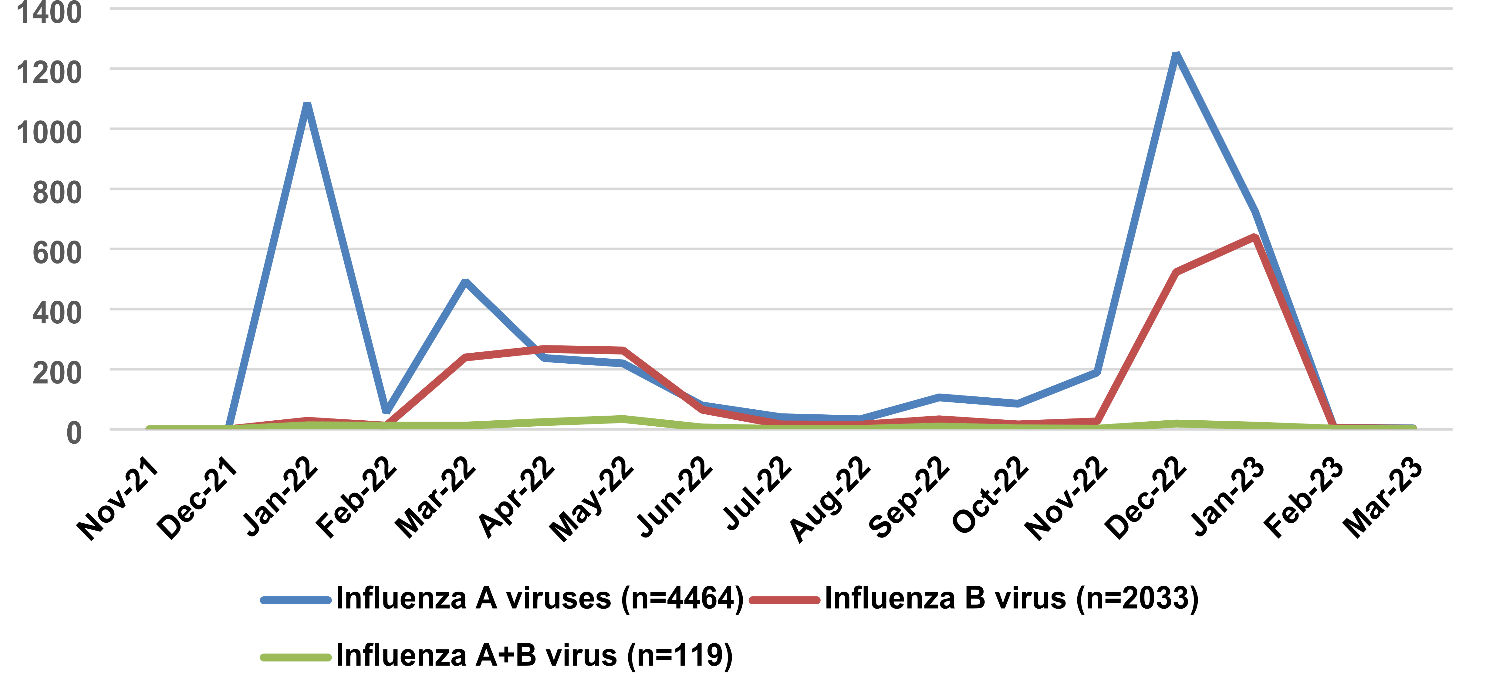
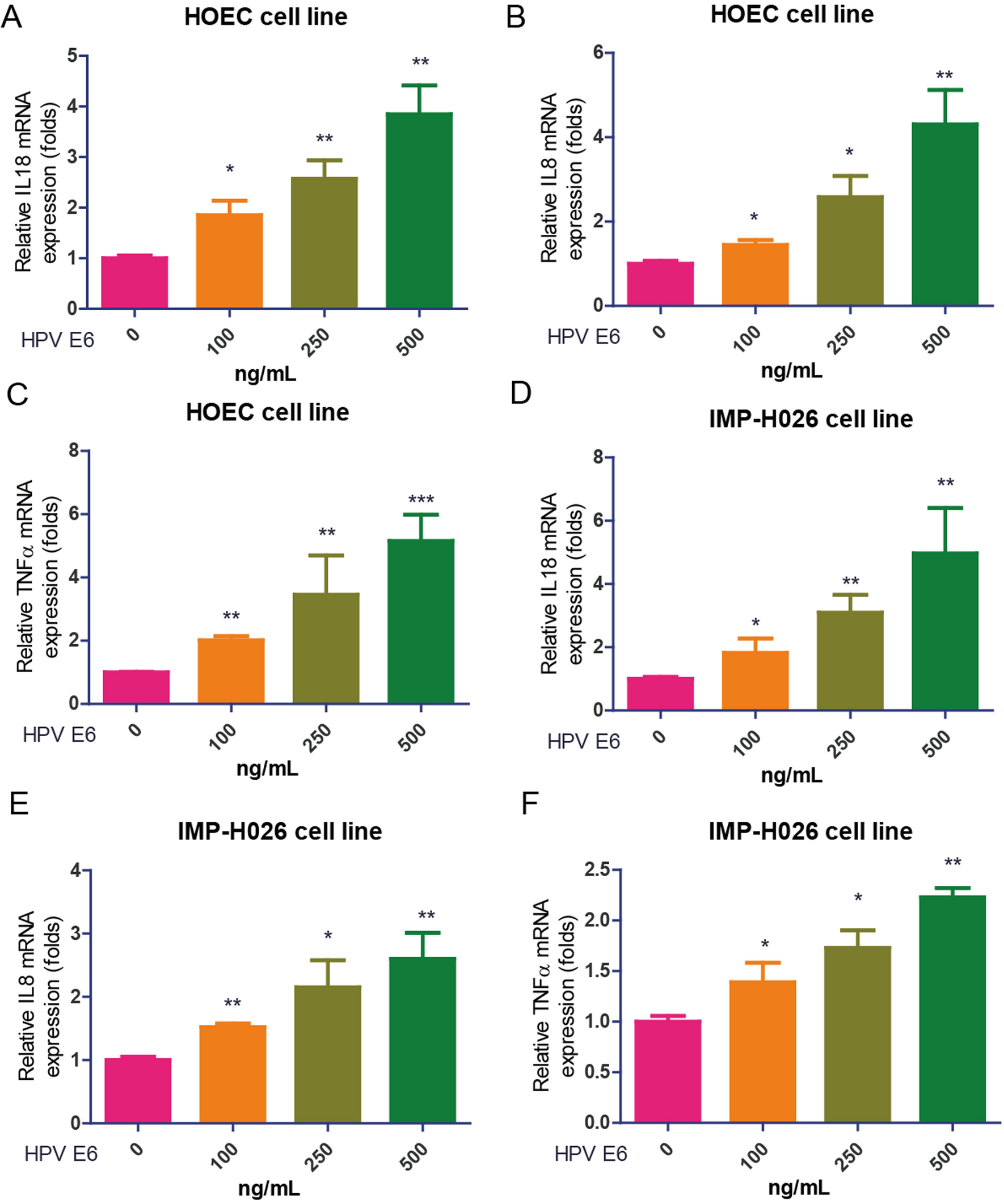
記住我




The normal IPEC-J2 cells exhibited irregular shapes with clear outlines and distinct boundaries and were evenly distributed on the cell plate. After infection with the classical PEDV strain CV777, the cells showed obvious shrinkage, became rounded, and lost their normal cellular morphology, indicating cytopathic effects typical of PEDV infection (Fig. 1A). The qPCR analysis revealed that the copy number of the PEDV-M gene was increased at 12 h, reached the highest expression level at 24 h, and was slightly decreased at 48 h postinfection (Fig. 1B).
Fig. 1
PEDV infection induces damage to IPEC-J2 cells at different time points. (A) Observation under an optical microscope. (B) PEDV copy numbers in IPEC-J2 cells at different time points postinfection. Data are presented as the mean ± SD, n = 3. Different letters indicate significant differences, P < 0.01
Transcriptome sequencing was subsequently performed on samples from different time points. A relatively uniform distribution of TPM values for all sample genes was observed (Fig. 2A). The PCA results also indicated that the genes within each group presented similar expression patterns, while the samples in the different groups were well distinguished (Fig. 2B). These results suggested that the sequencing data quality was satisfactory for further bioinformatics analysis. Differential analysis revealed 2,275 (611 upregulated and 1,664 downregulated), 1,492 (609 upregulated and 883 downregulated), 3,409 (2,093 upregulated and 1,316 downregulated), 2,231 (1,509 upregulated and 722 downregulated), 5,417 (3,398 upregulated and 2,019 downregulated), and 2,703 (1,951 upregulated and 752 downregulated) DEGs in the 12 h vs. Mock, 24 h vs. Mock, 48 h vs. Mock, 24 h vs. 12 h, 48 h vs. 12 h, and 48 h vs. 24 h comparison groups, respectively (Fig. 2C, D).
Fig. 2
Analysis of differentially expressed genes. (A) Box plot of log2(TPM) values for mRNAs across different time points. (B) PCA diagram of normalized mRNA expression values illuminating the general relationships among datasets. (C) Differential gene expression results showing up- and downregulated genes in the six comparison groups (12 h vs. Mock, 24 h vs. Mock, 48 h vs. Mock, 24 h vs. 12 h, 48 h vs. 12 h and 48 h vs. 24 h). The threshold used to define the DEGs was a |log2(FC)| >1 and an adjusted p value < 0.05. The blue dots indicate downregulated genes, and the red dots indicate upregulated genes. (D) Histograms of the number of differentially expressed genes
Enrichment analysis revealed that pathways such as the JAK-STAT signaling pathway, MAPK signaling pathway, cytokine‒cytokine receptor interaction, and PI3K‒Akt signaling pathway were enriched in the Mock group. Immune-related pathways, including regulation of the T-cell apoptotic process, regulation of lymphocyte differentiation, and regulation of the adaptive immune response, were enriched only in the 48 h vs. Mock comparison. Pathways such as carbohydrate metabolism, the HIF-1 signaling pathway, and positive regulation of lipid transport were enriched exclusively in the 12 h vs. Mock comparison (Fig. 3).
Fig. 3
GO, KEGG, and REACTOME enrichment analyses. The y-axis represents pathway entries, and the x-axis represents the grouping of differentially expressed genes. The shape of the plot represents different databases, with circles representing the GO database, triangles representing the KEGG database, and squares representing the REACTOME database. The color represents the magnitude of the p value, with redder color indicating a smaller p value. Group Specific: pathways enriched only in the differential genes of a single group. Group Two: pathways enriched in the differential genes of exactly two groups. Group Wide: pathways enriched in the differential genes of exactly three groups
Time-course transcriptomics uncovers transcriptional reprogramming during PEDV infectionTo investigate the gene expression trends across different groups, we employed a soft-threshold clustering method based on the within-group sum of squares “gap” statistic (Fig. 4A) and classified the gene expression patterns into 5 clusters (Fig. 4C and Table S1). A gene expression heatmap was constructed to display the expression patterns of genes from Cluster 1 to Cluster 5 (Fig. 4B). The expression of genes in Cluster 1 tended to increase after PEDV infection, and enrichment analysis revealed that these genes were involved mainly in antiviral pathways, such as the defense response to viruses, regulation of immune system processes, and TNF signaling. Conversely, genes in Clusters 3 and 4 tended to be downregulated following PEDV infection and were enriched primarily in metabolism-related pathways, including the cellular amide metabolic process, liposaccharide metabolic process, and carbohydrate derivative metabolic process. Cluster 2 and Cluster 5 presented irregular expression patterns over time and were enriched mainly in pathways such as RNA splicing, rRNA processing, the cell cycle, and tight junctions (Figs. 4D and 5A and B and Tables S2, 3).
Fig. 4
Global changes in gene expression across multiple time points. (A) Analysis plot for determining the optimal number of gene clusters on the basis of the total within-cluster sum of squares and the “gap” statistic. (B) Heatmap of genes highlighting the different expression patterns. The colors on the y-axis indicate the different gene expression pattern clusters, whereas the colors on the x-axis indicate the different sample groups. The intensity of the heatmap colors indicates the relative expression levels, with red representing increased expression and blue representing decreased expression. (C) Line plots showing the average gene expression trends for different expression patterns. (D) Line plots of the differential gene expression patterns. The line colors represent the membership of genes in the clusters, with darker red indicating stronger membership and darker blue indicating weaker membership in the respective cluster
Fig. 5
Enrichment analysis of different gene expression pattern clusters. (A) Enrichment analysis based on the GO database. (B) Enrichment analysis based on the KEGG database. The x-axis represents the different gene expression pattern clusters, and the y-axis indicates the pathway entries. The color of the points also indicates the gene expression pattern clusters. The size of the points represents the number of genes in the indicated pathway, with larger points indicating a greater number of genes
WGCNA identifies time-specific coexpression modules in PEDV infectionThis study utilized WGCNA to investigate gene coexpression networks associated with the PEDV infection process. The sample clustering tree revealed no outlier samples, indicating that all the samples could be further analyzed via WGCNA (Fig. 6A). With the soft threshold β set to 14 (Fig. 6B), WGCNA successfully divided the genes into eight coexpression modules represented by different colors (Fig. 6C), with each module exhibiting distinct expression patterns (Fig. 6D). Among these modules, the brown gene module (r=-0.64, p = 8e-03), cyan gene module (r = 0.76, p = 7e-04), dark red gene module (r=-0.85, p = 3e-05), and dark gray gene module (r = 0.72, p = 2e-03) were significantly correlated with the PEDV infection process (Fig. 6E) (Tables S4, S5, S6 and S7).
To further explore the functions of the gene modules significantly associated with the PEDV infection process, we performed enrichment analysis on the key genes (MM ≥ 0.8, GS ≥ 0.8) within the brown, cyan, dark red, and dark gray gene modules (Fig. 6F). GO enrichment analysis revealed that these genes were involved primarily in biological pathways such as T-cell proliferation, positive regulation of cell–cell adhesion, and regulation of cell proliferation (Fig. 7A). KEGG enrichment analysis revealed enrichment of pathways including the TNF signaling pathway, viral protein interaction with cytokines and cytokine receptors, the Toll-like receptor signaling pathway, and the JAK-STAT signaling pathway (Fig. 7B). Reactome enrichment analysis revealed enrichment in biological pathways such as extracellular matrix organization, homology-directed repair, and KK complex recruitment mediated by RIP1 (Fig. 7C).
Fig. 6
WGCNA identified gene coexpression modules at different time points after PEDV infection. (A) Sample clustering tree at different time points postinfection. (B) Selection of the soft-thresholding power (β). The left panel displays the scale-free fit index in relation to the soft-thresholding power. The right panel shows the mean connectivity versus the soft-thresholding power. A power of 14 was chosen because the fit index curve flattens out at higher values (> 0.8). (C) Hierarchical cluster dendrogram of samples at different time points postinfection, showing the coexpression modules generated via WGCNA. Modules belonging to branches are color-coded according to the interconnectedness of genes. Eight modules represented by colors in the horizontal bar were found via a 0.25 threshold for merging. (D) Heatmap showing the gene expression of the 8 modules across the four time points. (E) Relationships between modules and time points after PEDV infection. The correlation heatmap displays the correlations between modules, with the color intensity representing the strength of the correlation. A deeper red color indicates a stronger positive correlation between modules, whereas a deeper blue color indicates a stronger negative correlation. The lines connecting the time points and the 8 modules represent the associations between the traits and the coexpression modules. The line thickness represents the strength of the association, with thicker lines indicating stronger associations. The line colors represent the p values of the associations, with deep red for p < 0.01, orange for 0.01 < p < 0.05, and gray for p > 0.05. (F) Scatter plot of gene significance vs. module membership for modules associated with different postinfection time points. The y-axis represents gene significance, which indicates the degree of association between a gene and the infection time point trait. Genes with high significance may have particularly important biological functions or regulatory roles under specific phenotypic conditions. The x-axis represents module membership, with higher values indicating that a gene expression pattern is more closely correlated with the overall module. The color of the points corresponds to the four modules associated with each time point (deep red, cyan, brown, and dark gray)
Fig. 7
Enrichment analysis of genes within coexpression modules associated with PEDV infection time. (A) Bubble plot of the GO enrichment analysis results. (B) Bubble plot of the KEGG enrichment analysis results. (C) Bubble plot of the Reactome enrichment analysis results. The x-axis represents the enrichment factor, which is the ratio of the number of DEGs in a pathway to the total number of genes in that pathway. The size of the dots represents the number of genes, with larger dots indicating a greater number of genes. The color of the dots represents the magnitude of the p value
PPI network analysis reveals regulatory hubs in PEDV infectionTo identify key core regulatory factors during the PEDV infection process, we conducted protein interaction network analysis. Among the 985 key genes associated with PEDV infection, the top 20 core genes in the interaction network included 6 upregulated genes (TFRC, SUOX, RMI1, CD74, IFIH1, and CD86) and 14 downregulated genes (FOS, CDC6, CDCA3, PIK3R2, TUFM, VARS, ASF1B, POLD1, MCM8, POLA1, CDC45, BCS1L, RAD51, and RPA2) (Fig. 8A, B). GSEA of the 48 h vs. Mock group revealed that among the 20 core genes, the only enriched genes were RAD51, CDC6, and RPA2, which participate primarily in pathways such as DNA replication and homologous recombination (Fig. 8C, D).
Fig. 8
Protein interaction network revealing core genes and GSEA enrichment analysis. (A) Protein–protein interaction network. The size of the nodes represents the importance of the genes in the protein interaction network, with larger nodes indicating proteins with more interaction partners. The top 20 core genes are labeled. (B) Heatmap of the top twenty core genes. The colors of the horizontal axis represent different sample groups. The depth of color in the heatmap represents the level of expression, with red indicating high gene expression and blue indicating low gene expression. (C-D) GSEA enrichment analysis plot. The x-axis represents the ranking of gene sets, with the ranking decreasing from left to right, whereas the y-axis represents the enrichment score. The enrichment score curve shows the degree of enrichment of a gene set during the PEDV infection process, with a larger absolute value of the curve peak indicating a greater degree of enrichment of the gene set during the PEDV infection process. (E) Bar plot of the fold change in core gene RNA-seq and qPCR expression. The y-axis shows the log2FoldChange, and the x-axis indicates the analyzed gene. Data are presented as the mean ± SD, n = 3
留言 (0)