
記住我




Specimens from seven MPXV patients were collected in the Nantong area of Jiangsu, China, between September 11, 2023, and January 24, 2024. The results of the epidemiological investigation revealed that all seven patients in this study were male and were diagnosed with HIV. Except for one patient whose source of infection remains unclear, all other patients engaged in male-to-male sexual behavior within 21 days prior to the onset of symptoms, indicating a pattern of sexual transmission among men who have sex with men. As shown in Table 1, none of the seven patients had a history of travel or residence abroad. Two of the cases lived together and both visited a high-risk bathhouse in Shanghai, while the other patients were not linked to each other.
Table 1 Summary and organization of patient epidemiological dataSequencing results of MPXVThis study utilized the Illumina sequencing platform for whole-genome sequencing, with a minimum output of 4000 K reads or 600 M bases per sample. The average data output for each sample was 2 Gb, and one sample failed to produce sequencing data due to insufficient library concentration. The Q30 value for each sample exceeded 94%. After obtaining the sequencing data, a 10 × sequencing depth was selected to generate the final genome sequence. Utilizing NC_063383 as the reference sequence, the sequencing data were aligned, resulting in six complete MPXV genomes with sequence lengths ranging from 197,137 to 197,152 bp, as presented in Table 2. The six genomic sequences were annotated using Geneious Prime and subsequently submitted to the National Microbial Science Data Center (registration numbers: NMDCN0005SPG, NMDCN0005SPH, NMDCN0005SPI, NMDCN0005SPJ, NMDCN0005SPK, and NMDCN0005SPL).
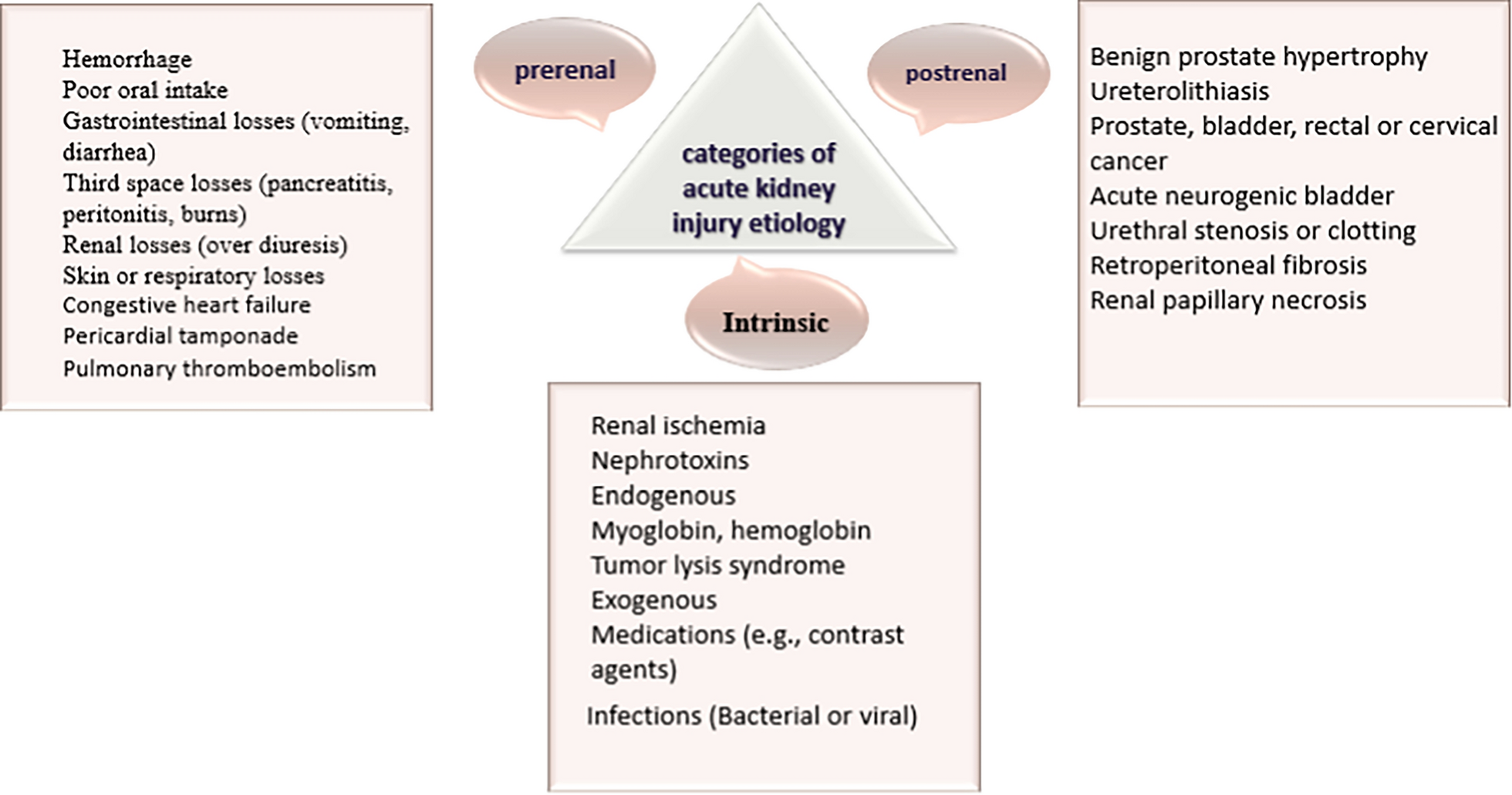
Table 2 Compilation of MPXV whole genome sequencing output dataPhylogenetic characteristics of MPXV in Nantong, JiangsuTo gain insights into the sources of MPOX cases reported between September 2023 and January 2024, an analysis of six MPXV genome sequences was conducted using the online tool Nextclade v3.8.2. The dataset for MPXV, which includes all clades, consists of 567 genome sequences from 42 countries across six continents, along with the six MPXV genome sequences from Nantong, Jiangsu, for phylogenetic analysis. The findings reveal that all six MPXV genome sequences are classified within Clade IIb, Lineage C.1.1 (as shown in Fig. 1).
Fig. 1
Phylogenetic Tree Analysis of MPXV Samples from Nantong, Jiangsu, and Other Global Data
This phylogenetic tree, constructed using whole-genome SNP (single nucleotide polymorphism) data, illustrates the phylogenetic relationships among 567 strains of MPXV. In this tree diagram, branches are colored to represent different clades by Nextclade v3.8.2, with the background color of each branch indicating different lineages. The two outer rings of the tree represent “Continent” and “Country of Origin.” The ring closest to the sequence ID indicates "Continent," while the outermost ring represents "Country of Origin," with labels provided next to the rings. The six sequences from this study are highlighted in text.
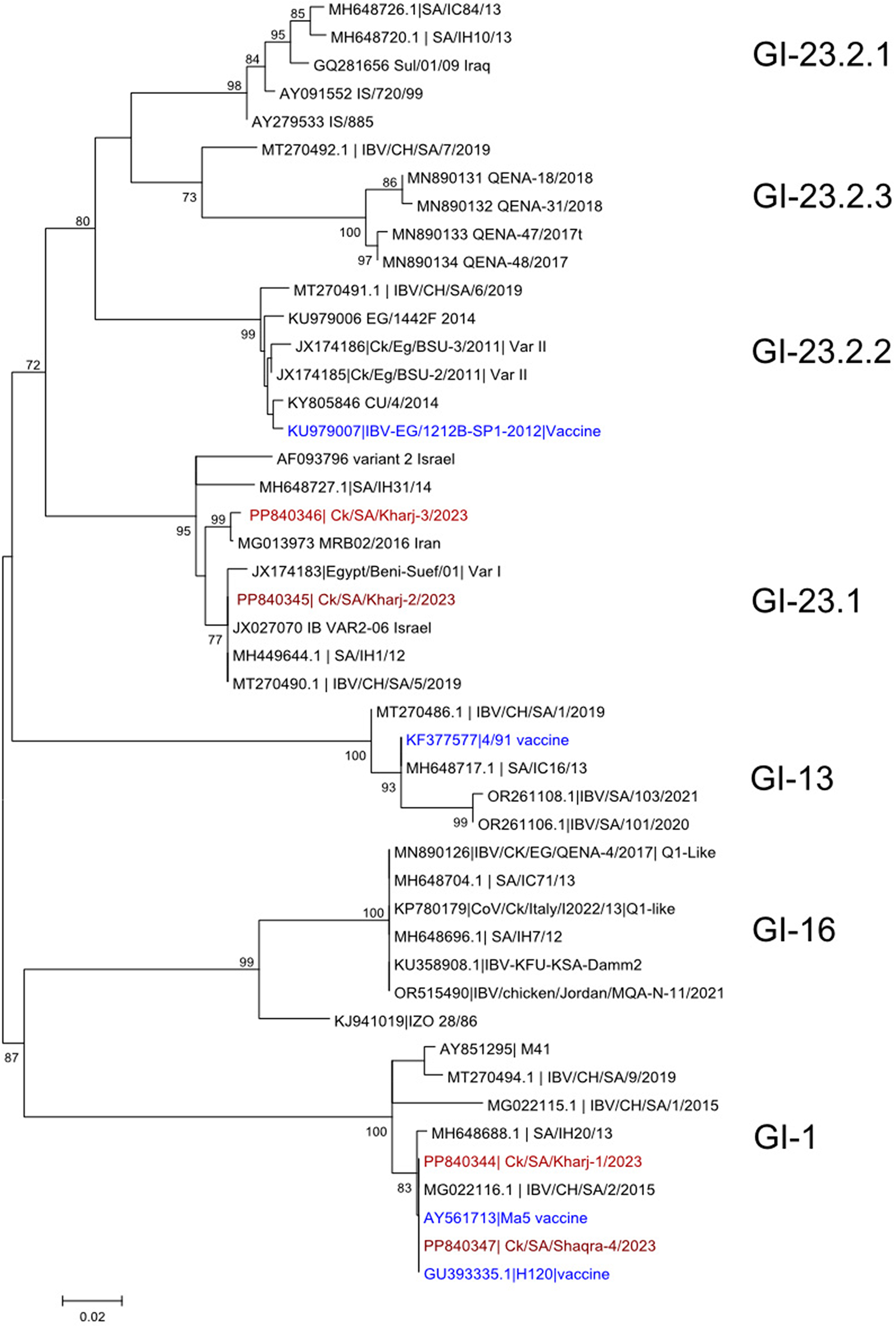
Phylogenetic characteristics analysis of lineages C.1 and C.1.1To trace their origins, this study presented sequences from the C.1 and C.1.1 lineages in Asia and Europe, sourced from GISAID and NCBI, along with sequences of the C.1.1 lineage identified in Shenzhen from the National Microbial Science Data Center, totaling 94 genomes. These sequences were utilized to construct phylogenetic trees for the C.1 and C.1.1 lineages. The analysis revealed that the six cases in this study are distributed across three distinct genetic clusters, suggesting that they may have originated from three different transmission chains. Notably, the genome sequences of three cases from Nantong, Jiangsu (IDs: NMDCN0005SPJ, NMDCN0005SPG, NMDCN0005SPL) corresponded with those of two cases in Germany (NCBI: OR777677.1 and PP093713.1). Additionally, the genome sequences of a case in the United States (NCBI: PP507063.1) and a case in Beijing, China (GISAID ID: EPI_ISL_18257122) were found within the same sequence cluster. Based on evolutionary distance and onset time, the MPXV sequences from the three cases in Nantong, Jiangsu, and the two cases in Germany exhibited a close relationship. The genome sequences of two cases from Nantong, Jiangsu (IDs: NMDCN0005SPH and NMDCN0005SPK) formed a separate cluster. Notably, the genome sequence of a case from Nantong, Jiangsu (ID: NMDCN0005SPI) is isolated in its own cluster but is closely related to a case from Shenzhen (ID: C_AA061331.1) as well as four cases from Japan (GISAID IDs: EPI_ISL_18352305, EPI_ISL_18352303, EPI_ISL_18352306, EPI_ISL_19001887). Furthermore, the genome sequences of one case from the United States (NCBI: OR643705.1) are also found within the same cluster. Phylogenetic tree analysis suggests these three distinct genome clusters may have originated from a case in Japan (GISAID ID: EPI_ISL_17692269) (as shown in Fig. 2).
Fig. 2
Phylogenetic Analysis of 92 MPXV Genomes from Lineages C.1 and C.1.1, Including Nantong Cases
Phylogenetic Tree Construction for Lineages C.1 and C.1.1 Based on Multiple Sequence Alignments. The background color on the phylogenetic tree represents different lineages. In this tree, “Location” denotes the country, “City” denotes the city (China), “ID” denotes the registration number of the six cases from Nantong, “Lineage” indicates the pedigree, and "Date “represents the time of virus genome acquisition. The location, city and date were labeled in the right of tree. The sequence of lineage B.1.3 (ID: OP480420.1) was selected as the outgroup, indicated by a green background color.
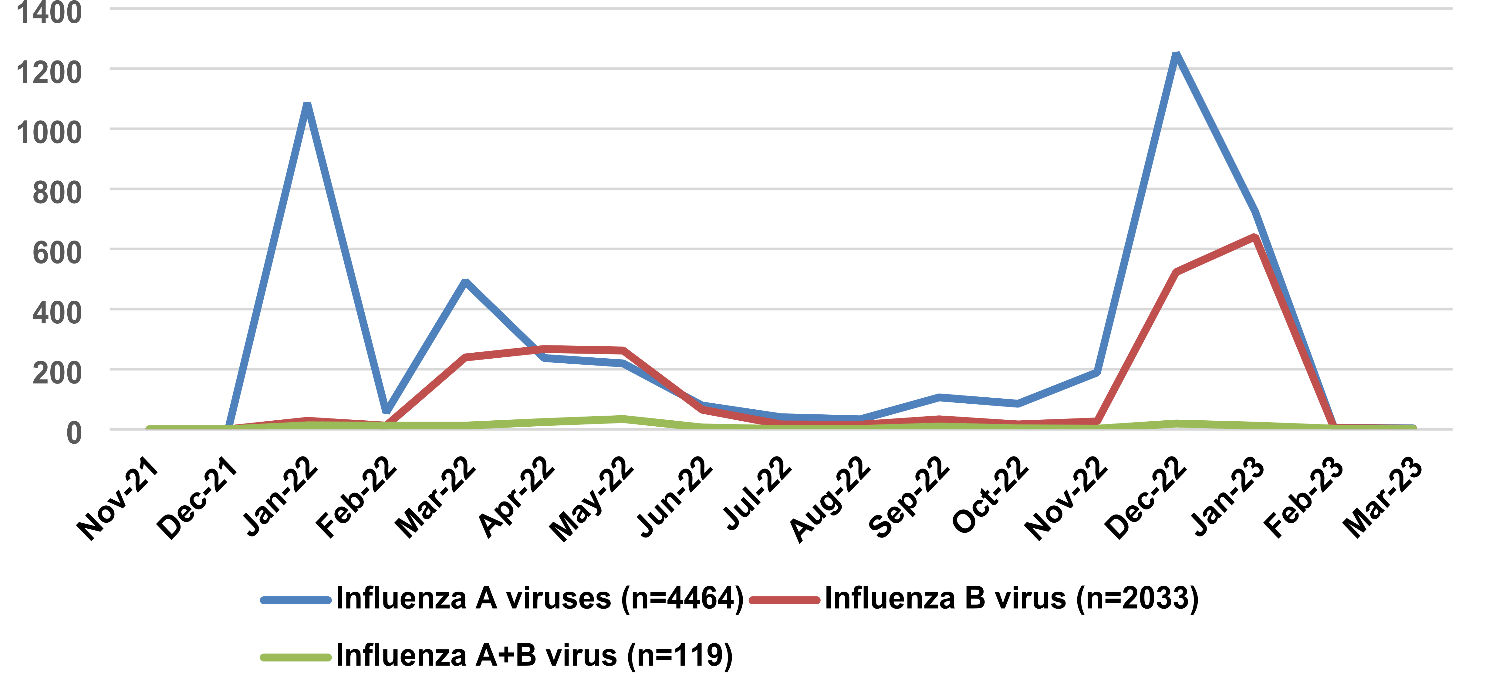
Evolutionary mechanisms of lineage C.1.1 in Nantong, Jiangsu provinceMicroevolutionary analysis involves the examination of minor genetic variations within viral genomes, particularly focusing on single or a few nucleotide mutations. Mutations mediated by the APOBEC3 enzyme are widely recognized as significant contributors to the evolutionary dynamics and adaptive modifications of MPXV, characterized by specific dinucleotide mutations such as GA → AA and TC → TT [28]. In this study, we selected 12 sequences from the C.1 and C.1.1 lineages shown in Fig. 2, using MT903344.1 as the reference sequence, and performed a specific locus analysis with six local sequences. The results revealed that these six sequences contained 16 unique loci distinct from those in other lineages. The detailed results are shown in Fig. 3a.
Fig. 3
SNP analysis of six MPXV genomes from Nantong, Jiangsu
To further investigate the mutation characteristics of these six genomic sequences, we used the Snipit package to efficiently extract and visualize the variant sites in the six aligned MPXV genomic sequences. After analysis and compilation, we found that these six MPXV genomic sequences exhibited 64, 61, 58, 66, 63, and 64 mutations, respectively, with the number of non-synonymous mutations being 29, 28, 27, 31, 30, and 29. In addition, Python scripts were used in this study to further efficiently screen for APOBEC3-like mutations in these variants. The results indicated that in this study, the six MPXV genomic sequences displayed five APOBEC3-like mutations of GA → AA and three of TC → TT, with the majority of mutations being non-APOBEC3 enzyme-mediated as shown in Fig. 3b.
These SNPs were extracted from the Jiangsu Nantong MPXV through multiple sequence alignment with the reference sequence (ID: MT904433.1). In Fig. 3a, compared to the reference sequence MT903344.1, the six local genomic sequences showed some unique mutation sites when compared to sequences from the C.1 and C.1.1 lineages. The light blue background represents C.1 lineage sequences, the dark blue background represents the six genomic sequences from this study, and the light green background represents other C.1.1 lineage sequences. Mutation sites in red font indicate unique mutations in the six genomic sequences from this study. In Fig. 3b, the mutation context refers to APOBEC3-like mutations, while the mutation types indicate synonymous mutations, nonsynonymous mutations, and other types of mutations. The pink and yellow boxes indicate APOBEC3-like mutations, while the green boxes represent synonymous mutations, and the blue boxes denote non-synonymous mutations.
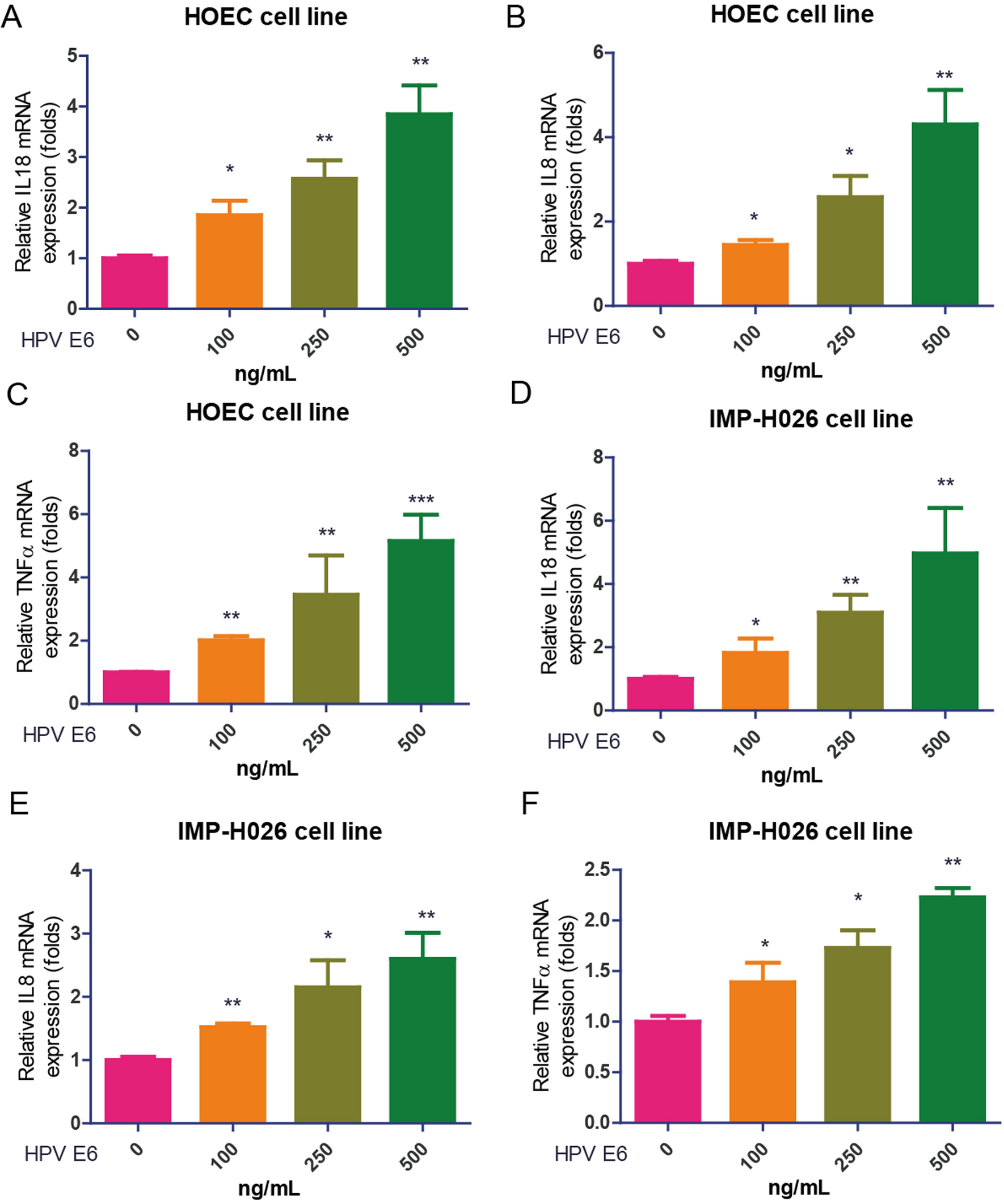
Protein changes induced by amino acid mutations in the C.1.1 lineageDuring the evolution of MPXV, alterations in protein function and structure may occur. To investigate the functional and structural changes of virus-related proteins, the modifications in viral proteins induced by SNPs were categorized. The results indicated that proteins associated with viral function have experienced mutations. For instance, proteins involved in host regulation and immune modulation include OPG003 (C19L, Ankyrin repeat protein), OPG0019 (D3R, Secreted growth factor), OPG42 (C3L, IFN resistance, PKR/eIF-alpha inhibitor), OPG25 (D9L, ankyrin-like protein), OPG37 (Cop-M1L-ANK-containing protein), and OPG176 (A47R, IL-1/TLR signaling inhibitor). Proteins related to viral replication and transcription comprise OPG93 (G9R, VLTF-1, late transcription factor), OPG125 (E12L, small subunit transcription initiation factor), OPG150 (A24R, VITF-3, intermediate transcription factor), OPG105 (J6R, DNA-dependent RNA polymerase), OPG145 (A19R, DNA helicase), and OPG180 (A50R, ATP-dependent DNA ligase). Additionally, other proteins primarily associated with viral particle assembly include OPG56 (C18L) and OPG64 (F2L), along with core viral proteins OPG115 (E3R) and OPG083 (I7L, similar to DNA topoisomerase II). Notably, most proteins linked to host regulation and immune modulation exhibit non-synonymous mutations, while proteins involved in viral replication, transcription, assembly, and maturation display a significant proportion of both mutation types (as shown in Fig. 4a). Furthermore, it was discovered that mutations in OPG36 (Cop-N2L), which exerts a strong inhibitory effect on RNA polymerase, can disrupt transcription initiation. The TNF receptor-like protein encoded by the virus, OPG002 (TNF receptor, CrmB), had its three-dimensional structural model predicted. The results show that the S54F mutation leads to a loss of hydrogen bonds, which may compromise the protein structure and affect its stability (as shown in Fig. 4b).
Fig. 4
Mutation abundance of different types of protein mutation and 3D Structural Variations in CrmB Protein
In Fig. 4a, the X-axis denotes specific amino acid mutation sites and their corresponding viral proteins, while the Y-axis reflects the frequency of these mutations. Synonymous mutations are indicated in yellow, whereas non-synonymous mutations are represented in green. Figure 4b presents the three-dimensional structural model of the CrmB protein, with ‘Wild-type’ denoting the structure of the amino acid site prior to mutation and ‘Mutant’ indicating the structure of the amino acid site following mutation.
留言 (0)