
記住我




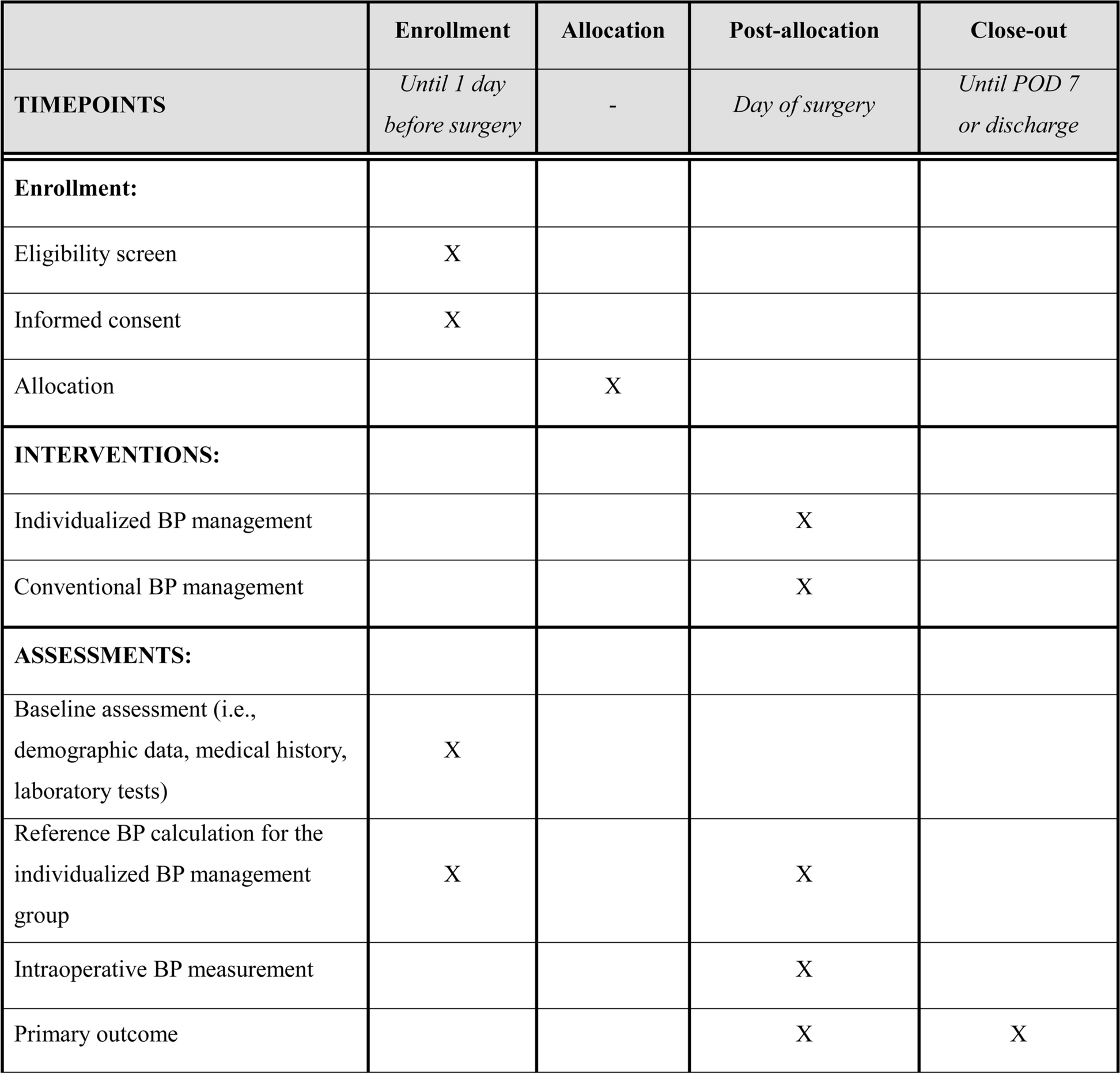


The proposed Seoul PeRioperative OUTcome research-4 (SPROUT-4) trial will be a multicenter, parallel-group, randomized controlled superiority trial of patients undergoing elective major noncardiac surgery. We plan to enroll patients from five tertiary university hospitals in South Korea. The study protocol was approved by the Institutional Review Boards of the five participating hospitals (Ajou University Medical Center, No. AJOUIRB-IV-2024–042 on January 22, 2024; Korea University Guro Hospital, No. 2024GR0016 on January 4, 2024; Samsung Medical Center, No. 2023–12-098–001 on February 15, 2024; Seoul National University Bundang Hospital, No. B-2402–883-402 on February 2, 2024; Seoul National University Hospital, No. 2312–127-1496 on January 10, 2024) and registered at ClinicalTrials.gov on January 26, 2024 (NCT06225453). The trial coordinating center will be Seoul National University Hospital. The proposed trial will be conducted in compliance with the guidelines for Good Clinical Practice and the Declaration of Helsinki [13]. This manuscript adheres to the Standard Protocol Items: Recommendations for Interventional Trials statement (Fig. 1 and Additional file 1) [14].
Fig. 1
Study schedule for enrollment, interventions and assessments. POD, postoperative day; BP, blood pressure
PatientsWe will include high-risk patients scheduled for elective major noncardiac surgery under general anesthesia with an expected duration of more than 2 h. The high-risk criteria were determined based on a literature review [15]. Patients at a high risk are defined as those aged ≥ 65 years or those aged ≥ 45 years with more than one of the following: coronary artery disease, peripheral vascular disease, transient ischemic attack or stroke, or congestive heart failure. We will exclude patients undergoing emergency surgery, organ transplantation surgery, or brain or carotid artery surgery; patients with an American Society of Anesthesiologists physical status of 5 or 6; pregnant women; patients with uncontrolled hypertension, defined as SBP ≥ 180 mmHg or diastolic blood pressure ≥ 110 mmHg; patients with an estimated glomerular filtration rate < 30 ml/min/1.73 m2 or undergoing renal replacement therapy; patients with acute decompensated heart failure; or patients with sepsis, shock, or ongoing inotrope or vasopressor infusion.
Informed consentThe Executive Committee, consisting of one investigator from each participating center, will be responsible for patient enrollment. After verification of the inclusion and exclusion criteria, written informed consent will be obtained from all patients. Patients will be informed about the study’s purpose and procedures and the expected harm and benefits of participation. The English version of model consent form is provided in Additional file 2. Patients will be notified that their participation is voluntary and that they are free to withdraw at any time without any disadvantage.
Randomization and blindingPatients who provide consent will be randomly assigned in equal numbers to either the individualized (intervention group) or conventional blood pressure management group (control group). Randomization will be conducted (R software, version 4.3.0; R Foundation for Statistical Computing, Vienna, Austria) by an independent research assistant using a block size of two, four, six, or eight and stratified by the hospital. Access to the randomization sequence will be secure, requiring a password, and will be revealed on the day of surgery. Although the nature of the interventions means that attending anesthesiologists will inevitably know each patient’s group assignment, the patients, surgeons, ward physicians, and statisticians analyzing the study data will not know which group they belong to.
ProtocolA flowchart of the SPROUT-4 trial is shown in Fig. 2. The SPROUT-4 trial will not enforce specific restrictions on surgical procedures and perioperative management outside of the target blood pressure goals, thereby granting discretion to each participating hospital and attending anesthesiologist as well as closely mirroring real-world clinical practices. Consequently, there are no constraints on blood pressure measurement techniques (e.g., invasive arterial catheterization or noninvasive oscillometry), their application sites, measurement intervals, or the strategies implemented for maintaining designated blood pressure targets, including fluid and vasopressor administration, patient positioning, and anesthetic depth adjustment.
Fig. 2
Flowchart of the SPROUT-4 multicenter, randomized controlled trial. SBP, systolic blood pressure; MAP, mean arterial pressure; PACU, post-anesthesia care unit; ICU, intensive care unit
For the control group, the protocol dictates maintaining a MAP of at least 65 mmHg and an SBP of at least 90 mmHg throughout surgery for all patients. In the intervention group, MAP and SBP will be maintained at no less than 20% below baseline values. Baseline values will be calculated as the average of all MAP and SBP measurements obtained from the day before surgery until the morning of the surgery. These specified blood pressure targets are to be maintained until the patient is discharged from the post-anesthesia care unit or for those transferred to the intensive care unit after surgery until the end of surgery. For safety reasons, a minimum MAP of 50–55 mmHg will be maintained regardless of the calculated target values in the intervention group. This precaution ensures that blood pressure levels are kept within a safe range to mitigate the risk of hypotension-related adverse outcomes. If severe hypotension (defined as inability to maintain MAP above 50–55 mmHg despite interventions such as anesthetic adjustment, vasopressor administration, and fluid management) or any unexpected adverse events occur, the allocated intervention will be discontinued immediately, followed by appropriate therapeutic measures.
Outcomes and definitionsThe primary outcome will be a composite of all-cause death, stroke, myocardial infarction (MI), new or worsening congestive heart failure, unplanned coronary revascularization, and AKI. These outcomes will be assessed within 7 days after surgery or until discharge, whichever occurs first. The outcome components are defined as follows:
• Stroke: A new ischemic or hemorrhagic cerebrovascular accident with a focal neurological deficit confirmed using brain imaging
• MI: Diagnosed based on the Fourth Universal Definition of Myocardial Infarction (type 1, 2, or 3) [16]
▪ Type 1 MI
♦ Detection of a rise and/or fall of cardiac troponin levels with at least one value above the 99th percentile upper reference limit
♦ In addition, one of the following is required:
• Symptoms of acute myocardial ischemia
• New ischemic electrocardiography (ECG) changes
• Development of new pathological Q waves
• Imaging evidence of a new loss of viable myocardium or regional wall motion abnormality in a pattern consistent with an ischemic etiology
• Identification of a coronary thrombus by angiography, including intracoronary imaging or by autopsy
▪ Type 2 MI
♦ Detection of a rise and/or fall of cardiac troponin levels with at least one value above the 99th percentile upper reference limit
♦ Evidence of an imbalance between myocardial oxygen supply and demand unrelated to coronary thrombosis
♦ In addition, one of the following is required:
• Symptoms of acute myocardial ischemia
• New ischemic ECG changes
• Development of new pathological Q waves
• Imaging evidence of a new loss of viable myocardium or regional wall motion abnormality in a pattern consistent with an ischemic etiology
▪ Type 3 MI
♦ Cardiac death with symptoms suggestive of myocardial ischemia accompanied by presumed new ischemic ECG changes or ventricular fibrillation
♦ In addition, one of the following is required:
• Die before blood samples for biomarkers can be obtained
• Die before increases in cardiac biomarkers can be identified
• MI is detected by autopsy examination
• New or worsening congestive heart failure: Diagnosis on discharge letter of progression notes (medical records: pulmonary edema, congestive heart failure, etc.)
• Unplanned coronary revascularization: Percutaneous coronary intervention or bypass grafting, which was not an a priori planned stepwise procedure
• AKI is defined based on the serum creatinine criteria of the Kidney Disease: Improving Global Outcomes [17]: increase in serum creatinine level by 0.3 mg/dl or more within 48 h or to 1.5 times the baseline or more within 7 days. Baseline serum creatinine level is defined as the most recent preoperative value
The secondary outcomes will be individual components of the primary composite outcome, postoperative hospital length of stay, unplanned intensive care unit admission during index hospitalization, and new-onset atrial fibrillation of any duration, as captured by 12-lead ECG, continuous ECG monitoring, or telemetry, within 7 days after surgery or until discharge from the hospital, whichever occurs first.
Sample size calculation and statistical analysisBased on a pilot chart review conducted at the trial coordinating center (Seoul National University Hospital), the incidence of the primary outcome in the control group was estimated to be approximately 9%. We expected a 5% reduction in the incidence rate of the primary outcome in the intervention group. To detect this difference with an α of < 0.05 and a β of < 0.1 while assuming a dropout rate of 10%, we calculated that a total of 1896 participants, with 948 in each group, were necessary. There is no direct evidence in the literature regarding the expected 5% incidence of the primary outcome in the intervention group. However, we believe this is a conservative estimate. Should the benefits of this individualized approach prove significant, establishing such a conservative estimate ensures that any observed effects are robustly validated and attributable to the intervention itself, thereby minimizing the risk of overestimating its impact based on potentially confounding factors.
All analyses in the SPROUT-4 trial will be conducted in an intention-to-treat manner. Since the time window for all outcomes in this study is limited to the index hospitalization, complete follow-up is expected, with no anticipated missing data or loss to follow-up. Descriptive analyses will be performed to describe the baseline characteristics of the groups. The primary outcome will be compared between the groups using the chi-squared test. The relative risk and 95% confidence interval will also be reported. The same method will be applied to the binary secondary outcomes. The continuous secondary outcomes will be analyzed using Student’s t-test or Mann–Whitney U test, as appropriate. We will further compare the groups after excluding AKI from the primary composite outcome for sensitivity analysis. All analyses will be performed using the R software (version 4.3.0; R Foundation for Statistical Computing, Vienna, Austria). Statistical significance was set at P < 0.05.
Data management and data safety monitoringIn the SPROUT-4 trial, data management and oversight will be conducted to uphold integrity and confidentiality. Patients’ data will be collected by dedicated trial assistant at each participating hospital. Unique patient identifiers will facilitate anonymized data analysis. All pseudonymized data will be retained in strict compliance with pertinent data protection regulations. These personnel, blinded to group assignments, will manage access to the data through a secure, password-protected file, ensuring impartiality and integrity in the handling of trial data. Each trial site will implement oversight mechanisms to protect participant rights and wellbeing; guarantee the precision, completeness, and verifiability of trial data; and ensure adherence to the trial protocol, Good Clinical Practice, and relevant regulatory standards.
A Data and Safety Monitoring Board, consisting of two cardiovascular anesthesiology and perioperative care specialists, will convene every 6 months to rigorously review the trial data. Their responsibilities include evaluating patient safety (occurrence of severe hypotension defined as MAP < 50–55 mmHg despite interventions or any unexpected adverse events), monitoring trial progress, and ensuring data integrity. They may suggest amendments to the study protocol or advise on its continuation or termination, based on the results. Any proposed protocol amendments will undergo a comprehensive review and will require approval from the Institutional Review Boards of all participating hospitals prior to implementation in the clinical trial. In the case of an expected or unexpected serious adverse event with a reasonable causal relationship to the trial protocol, participants will be compensated for both the resulting harm and any additional medical expenses incurred.
DisseminationThe results of the SPROUT-4 trial will be reported to relevant scientific communities through publications in academic journals and presentations at national or international conference.
留言 (0)