
記住我
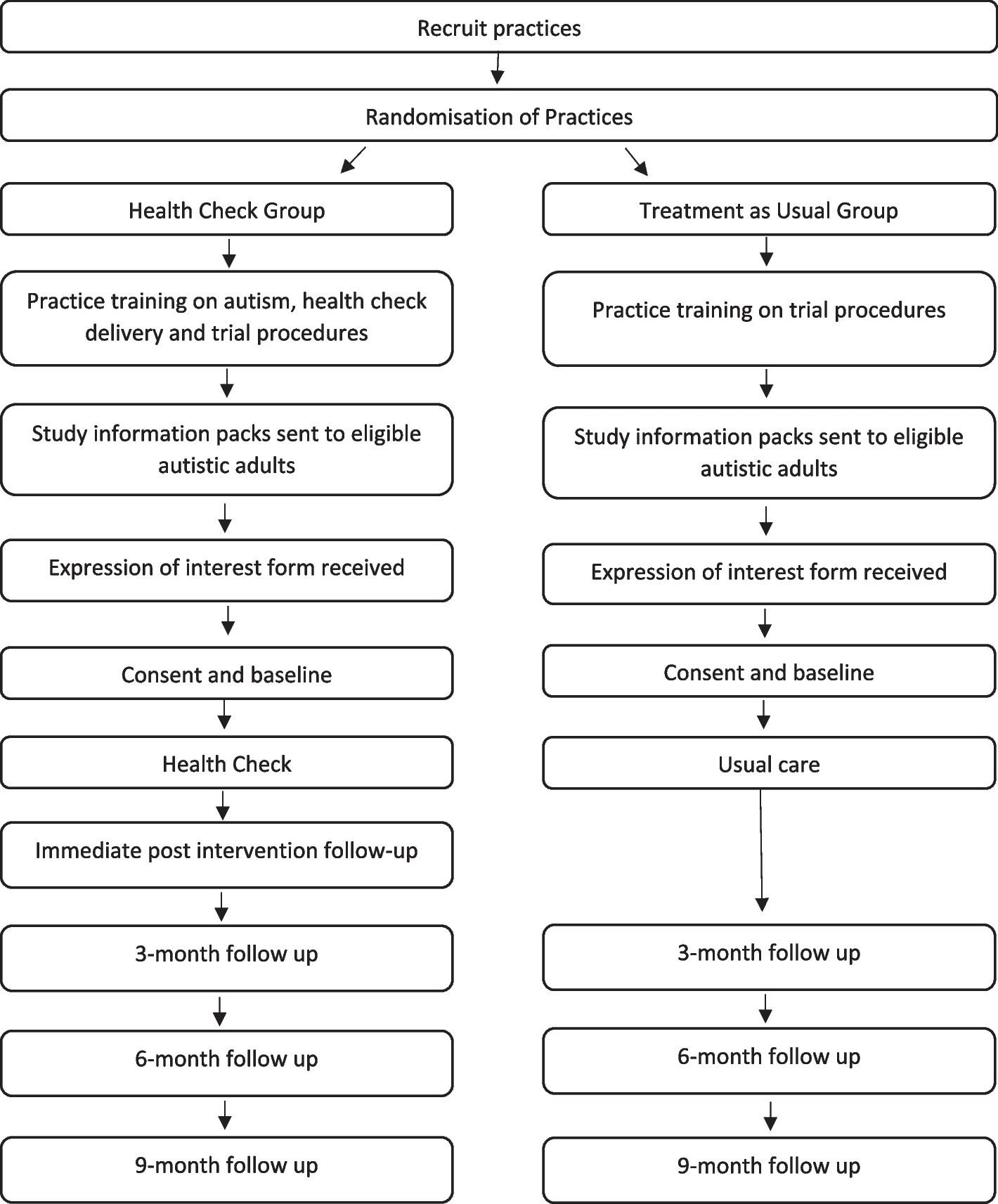




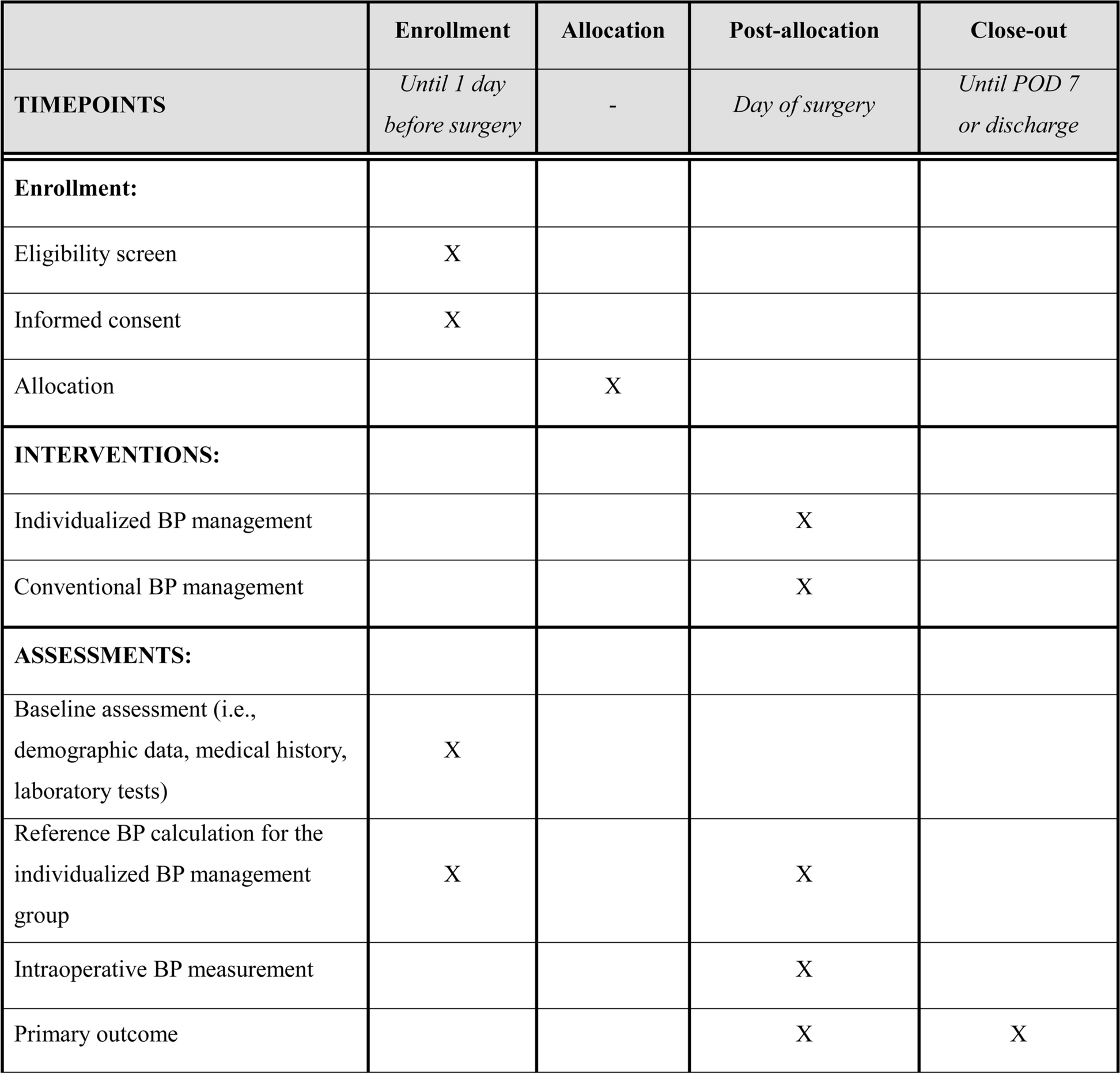


This multi-center, open-label, randomized clinical trial will be conducted at five hospitals in Nagoya, Japan (Nagoya University Hospital, Nagoya Central Hospital, Aichi Cancer Center, Japanese Red Cross Aichi Medical Center Nagoya Daiichi Hospital and Japanese Red Cross Aichi Medical Center Nagoya Daini Hospital), from February 2024 to December 2026. These hospitals were selected based on their high annual caseloads of craniotomies for brain tumors (exceeding 20 cases) and their comprehensive medical teams, which include at least three neurosurgeons and adequate support staff for intensive postoperative care. Each principal investigator at these sites is a neurosurgeon with over 20 years of experience as neurosurgeons, possess extensive knowledge, and expertise in managing brain tumors. Ethical approval was granted by the Ethics Committee of Nagoya University Hospital on December 7, 2023 (Approval number: 2023–0348). This study has been registered and published in the Japan Registry of Clinical Trials (jRCT) (Approval number: jRCTs041230117). The jRCT is an approved member of the Primary Registry Network of the World Health Organization’s International Clinical Trials Registry Platform. The study protocol adheres to the SPIRIT guideline [25]. Following SPIRIT’s recommendations, the schedule of enrolment, intervention, and assessment is summarized in Table 1, and a schematic representation of the study design is shown in Fig. 1.
Table 1 Timeline summary of enrolment, intervention, and assessments for the studyFig. 1
Schematic representation of the study design
Informed consentEach participant will receive a detailed description of the study from their investigator-in-charge and express their willingness to participate in the clinical trial by signing an institutional review board-approved consent form. The consent includes authorization for the collection and use of patient data and biological specimens in ancillary studies.
Patient selectionPatients will be selected based on the following inclusion and exclusion criteria, with only those treated according to standard clinical practice at the participating hospitals eligible for recruitment. Preclinical safety studies in patients without a history of epilepsy were conducted in adults, and this trial includes participants aged 18 years and older [26]. The upper age limit is set at 80 years to reduce the risks of AEs. A patient with “no history of seizures” in the inclusion criteria is defined as one who has no documented seizures and is not taking antiseizure drugs at the time of trial enrollment.
Inclusion criteriaPatients with supratentorial brain tumors scheduled for craniotomy
Patients with intra-axial tumors, extra-axial tumors with brain edema, or extra-axial tumors without brain edema compressing the motor cortex, diagnosed by the physicians in charge to be at high risk for early seizures after craniotomy
Karnofsky Performance Scale above 70
Ages from 18 to 80 years
No documented history of seizures
No contraindications to PER use
Written consent by the individual, a designated relation, attendant, next of kin, or surrogate
Exclusion criteriaPatients unable to take tablets orally
Patients indicated for awake surgery
Patients with central nervous system diseases other than brain tumors
Patients with a history of treatment for central nervous system diseases
Patients with a history of antiseizure drug use other than PER within 1 week of surgery
Patients deemed unsuitable for trial enrollment by the physicians in charge
Sample sizeThe sample size was determined using a one-sided significance level of 5%, a power of 80%, and an assumed dropout rate of 5%. We assumed an incidence rate of 20% in the control group and 5% in the PER group, requiring a total of 142 patients (71 per group). The assumed 20% incidence rate in the control group was based on the only prospective study examining the effect of antiseizure drugs on preventing early seizures after craniotomy, which reported an 18% incidence rate in the observation group [8]. For the assumed 5% incidence rate in the PER group, we referred to studies evaluating LEV, a relatively newer antiseizure drug, for seizure prophylaxis. In these retrospective studies, the incidence rate of early seizures after craniotomy ranged from 1.6 to 4.6% [12, 13]. Similarly, a retrospective study specifically investigating the prophylactic use of 2 mg PER reported a seizure incidence rate of 5% [17].
RandomizationPatients will be assigned (1:1) to either the treatment or control group using the minimization method in the web registration system and issued case numbers. Adjustment factors for allocation will be tumor type (primary brain tumor, metastatic brain tumor, or extra-axial tumor), sex, age (over or under 65 years), and institution. The physicians in charge will register and assign patients using this system.
Basic data collectionDemographic and clinical characteristics of the patients, including age, sex, medical history, family history of epilepsy, laboratory data, pre- and post-operative head magnetic resonance imaging reports, computed tomography findings, and pathological findings, will be recorded.
Treatment and measurementPatients in the treatment group will take 2 mg of PER tablets before sleep for 2 days prior to surgery and for 28 days after surgery. Eight 2 mg PER tablets are individually packaged on a single drug sheet. To facilitate patient compliance and adherence to the treatment protocol, patients will be instructed to continue taking the medication until day 28, the last day of the observation period. PER administration will be omitted on the day of surgery to minimize the risk of aspiration and account for variable dosing times due to surgery schedules. Postoperative blood PER levels will be measured. Patients in the control group will not receive any antiseizure drugs before or after surgery, as current guidelines do not recommend prophylactic antiseizure drug use in the perioperative period. If seizures occur in the PER group, the PER dose will be increased, or another antiseizure drug will be introduced at the discretion of the physicians in charge. In the control group, the antiseizure drugs use is prohibited until a seizure occurs. However, if a seizure does occur, an antiseizure drug will be administered according to standard clinical practice, with the specific drug choice left to the physician in charge. However, if a seizure occurs during the observation period, the patient will discontinue protocol treatment and any scheduled interventions.
Seizures will be diagnosed based on clinical manifestations, including involuntary movements, alterations in consciousness, or abnormal motor, sensory, or psychosensory phenomena. If the occurrence of a seizure is uncertain, an electroencephalogram will be performed, and a blinded adjudicating physician will make the final decision.
Postoperative follow-upDuring hospitalization, the physicians in charge will conduct daily safety assessments and monitor for seizure occurrences. Nurses will ensure that patients adhere to the prescribed PER tablet regimen by collecting empty drug sheets as evidence of compliance throughout the study. Blood tests for safety assessment will be performed on postoperative days 1, 7, 14, and 28. Patients may be discharged after day 7 if recovery is satisfactory. Upon discharge, an outpatient consultation will be scheduled for postoperative day 28, marking the final day of the protocol period, during which patients will undergo a blood test and submit empty drug sheets. Patients will also self-report their adherence to PER and their physical condition. Adherence during home care will primarily be based on patient self-reporting, ideally during outpatient visits, although telephone interviews or reports from next to kin are also acceptable. If a noticeable seizure occurs during home care, the physicians in charge will request the patient’s return to the hospital. For patients discharged between days 7 and 14, an additional outpatient consultation will be scheduled for postoperative day 14, where they will undergo a blood test, submit empty drug sheets, and self-report adherence to PER and physical condition.
Safety assessmentAEs will be recorded according to the Common Terminology Criteria for Adverse Events version 5.0. The number and incidence rates of AEs, along with the number of affected patients, will be summarized by AE type. Comparative analyses of AE incidence between the treatment and control groups will be conducted using Fisher’s exact test for each AE category. AEs will also be assessed for their relationship to the treatment (e.g., possibly, probably, or definitely related to PER). Serious adverse events (SAEs) will be reported separately. Regular monitoring will ensure patient safety and accurate data collection, as outlined in the protocol’s “Monitoring” section. Any grade 3 or higher AEs attributed to the treatment will be immediately reported to the monitoring committee for evaluation. Hematologic abnormalities (AST, ALT, γGTP) and clinical symptoms (dizziness, somnolence, irritability) potentially associated with PER will be recorded as grade 1 or higher, while all other AEs will be recorded as grade 3 or higher. Clinical symptoms will be documented based on patient-reported complaints from the initiation of PER administration. If a grade 3 or higher AE attributable to PER is identified, PER administration will be discontinued.
Handling of deathsAlthough deaths are not expected given the patient population and the nature of the intervention, the study protocol includes provisions for their occurrence. Any death occurring within the trial period will be thoroughly investigated to determine the potential association with the study intervention. Causes of death will be categorized (e.g., tumor progression, perioperative complications, or unknown). The incidence of death will be reported descriptively for each group. Suppose an unexpected death potentially related to PER occurs, it will be reported as a SAE and reviewed by the monitoring committee to ensure patient safety. Statistical analysis of mortality rates will not be conducted unless an unexpectedly high incidence of death occurs.
Planned outcomesThe primary outcome is the incidence of seizures within 28 days postoperatively. Secondary outcomes include the length of hospital stay, ICU stay, and incidence of postoperative complications.
Data managementResearchers will enter data collected during the observation period into an electronic data capture system (REDCap). The trial system was developed at a data center (Department of Advanced Medicine, Nagoya University Graduate School of Medicine). Data entered into REDCap will be monitored by the main data manager and maintained for 10 years post-study completion.
Data analysisAll collected data will be analyzed after the observation period for all cases by the main data-management manager, who will remain blinded to the treatment group. Patients who receive at least one dose of PER will be analyzed as the treatment group. The primary efficacy analysis population will be the Full Analysis Set (FAS), including all patients in the safety analysis population, excluding those with serious protocol violations (e.g., lack of consent, serious violations of study procedures) or without any post-treatment data. The primary analysis will follow the intention-to-treat principle, adhering closely to the FAS approach as recommended by ICH E9 guidelines. All patients who receive at least one dose of PER will be included in the treatment group analysis, regardless of adherence or protocol deviations. A per-protocol analysis will also be conducted as a secondary analysis to assess the efficacy of PER in patients who strictly adhered to the study protocol. These complementary analyses will strengthen the robustness of our findings.
For the primary endpoint, seizure incidence will be compared between groups using Fisher’s exact test within the FAS population. Hospital and ICU stays, measured from surgery day to discharge (with surgery day designated as day 0), will be analyzed using the Mann–Whitney U test. Postoperative complications, defined as surgery-attributable events, and their incidences will be analyzed using Fisher’s exact test. In case of missing, unused, or abnormal data, available data will be utilized. If substantial missing data is identified, exploratory analyses will be conducted to estimate its impact. A one-sided significance level of 0.05 will be used to test the superiority of the PER group over the control group for the primary endpoint. This study is designed as a superiority trial to assess the PER’s preventive effect on seizures after craniotomy. The primary objective is to demonstrate the PER’s superiority over the control group in reducing seizure incidence. Specifically, statistical significance will be declared if seizure incidence is significantly lower in the PER group compared to the control group. For all other analyses, a two-sided significance level of 0.05 will be used.
ConfidentialityIdentifying information about individual patients will be removed, and each patient will be assigned a research registration ID. A list linking the research registration IDs to the original pre-processing information will be maintained by the principal investigator on a network-disconnected computer. Data will be retained for 10 years post-study completion, accessible only to the principal investigator and the main data manager.
Plans to promote patient retention and complete follow-upPost-discharge medical interview appointments will be scheduled for all patients. If a patient does not attend the follow-up appointment, a telephone interview with the patient or family member will be conducted to gather the necessary information.
Composition of the coordinating center and the data management teamThe coordinating center for the clinical trial is located at Nagoya University Hospital consists of members from the Department of Neurosurgery, the Department of Advanced Medicine, and the Department of Pharmacy. This center is responsible for ensuring trial safety, verifying data accuracy, and distributing study drugs to other participating hospitals. The data management team, located within the Department of Advanced Medicine, handles system registration, data management, and final statistical analyses.
MonitoringA monitoring committee for this study has been established at Nagoya University Hospital. An individual who is not involved in the study and is accredited by Nagoya University Hospital is designated as the monitor. Monitors will conduct regular on-site or off-site monitoring and report the results to the principal investigator.
Interim analysisNo interim analysis is planned for this study. Therefore, the study will not be terminated based on interim findings.
AEs reportingAEs will be documented and assessed based on patient complaints and blood test results, as specified in the protocol. In the event of SAEs, physicians in charge will promptly provide necessary treatments and report them to the principal investigator. SAEs are defined as follows: death, life-threatening conditions, conditions requiring hospitalization or prolonged hospitalization for treatment, and permanent or serious disability/incapacity. For unintended SAEs, the principal investigator will report to the Pharmaceuticals and Medical Devices Agency.
Provisions for ancillary and post-trial careAny harms resulting from this clinical trial will be covered by clinical research insurance.
AuditingNo auditing is planned for this clinical trial.
Plans for communicating important protocol amendments to relevant partiesAll protocol amendments will be reviewed and approved by the Ethics Committee of Nagoya University Hospital. Approved amendments will be registered with the jRCT. The latest version of the protocol will be promptly distributed to all investigators.
Dissemination policyThe results of this clinical trial will be published in a peer-reviewed journal and made accessible to all interested parties. There are no plans to publish the full protocol or final dataset.
留言 (0)