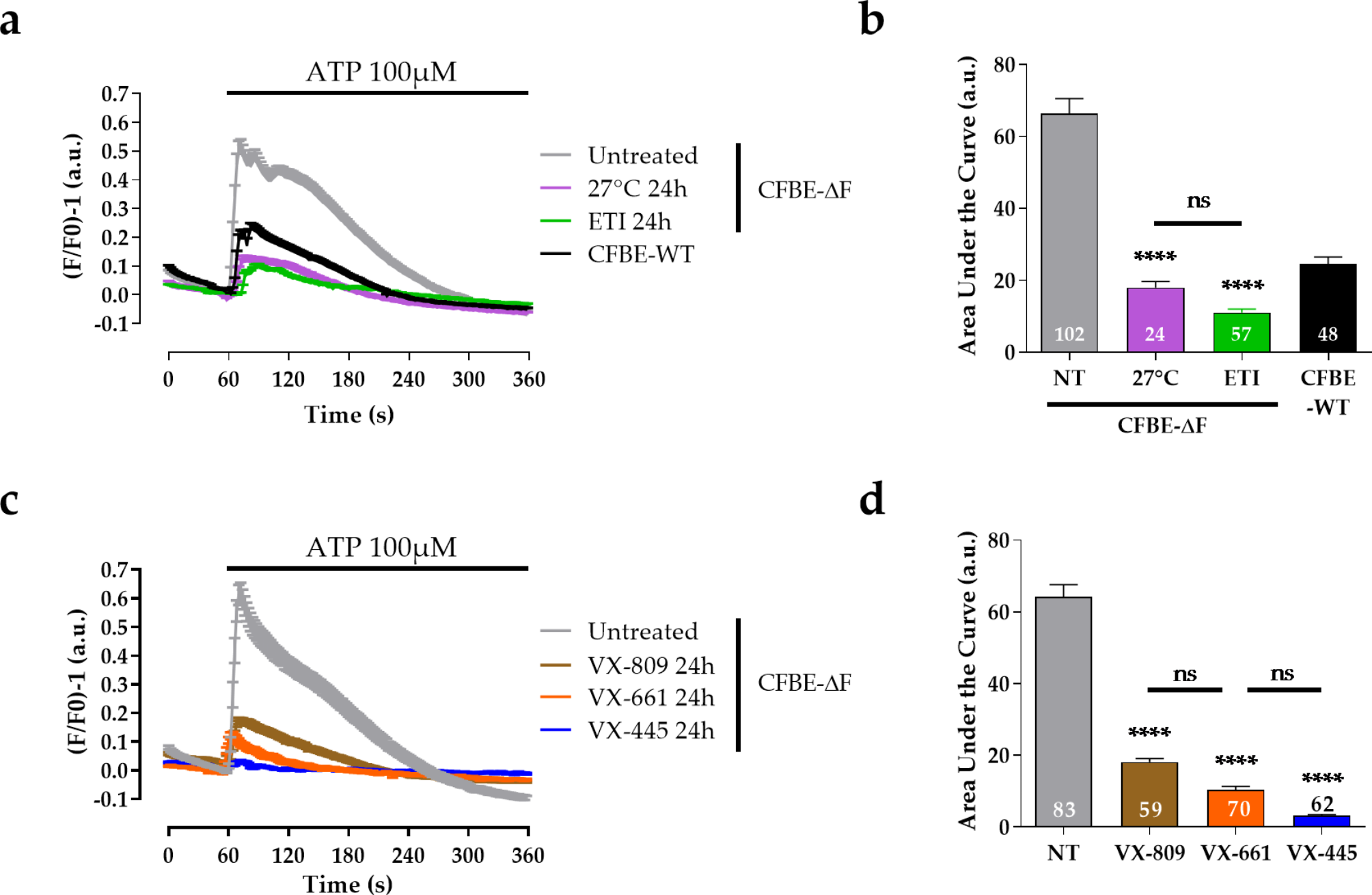
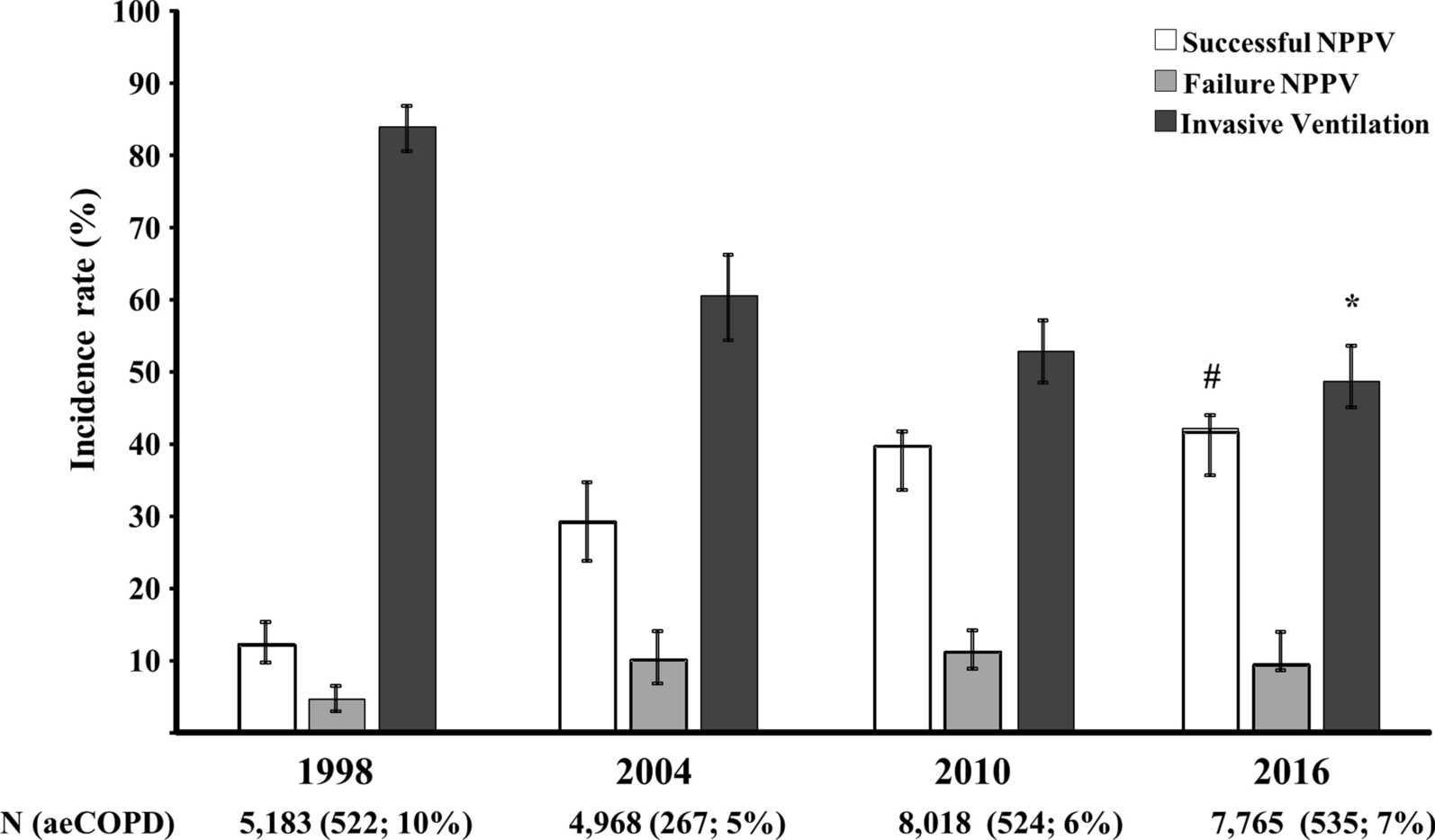
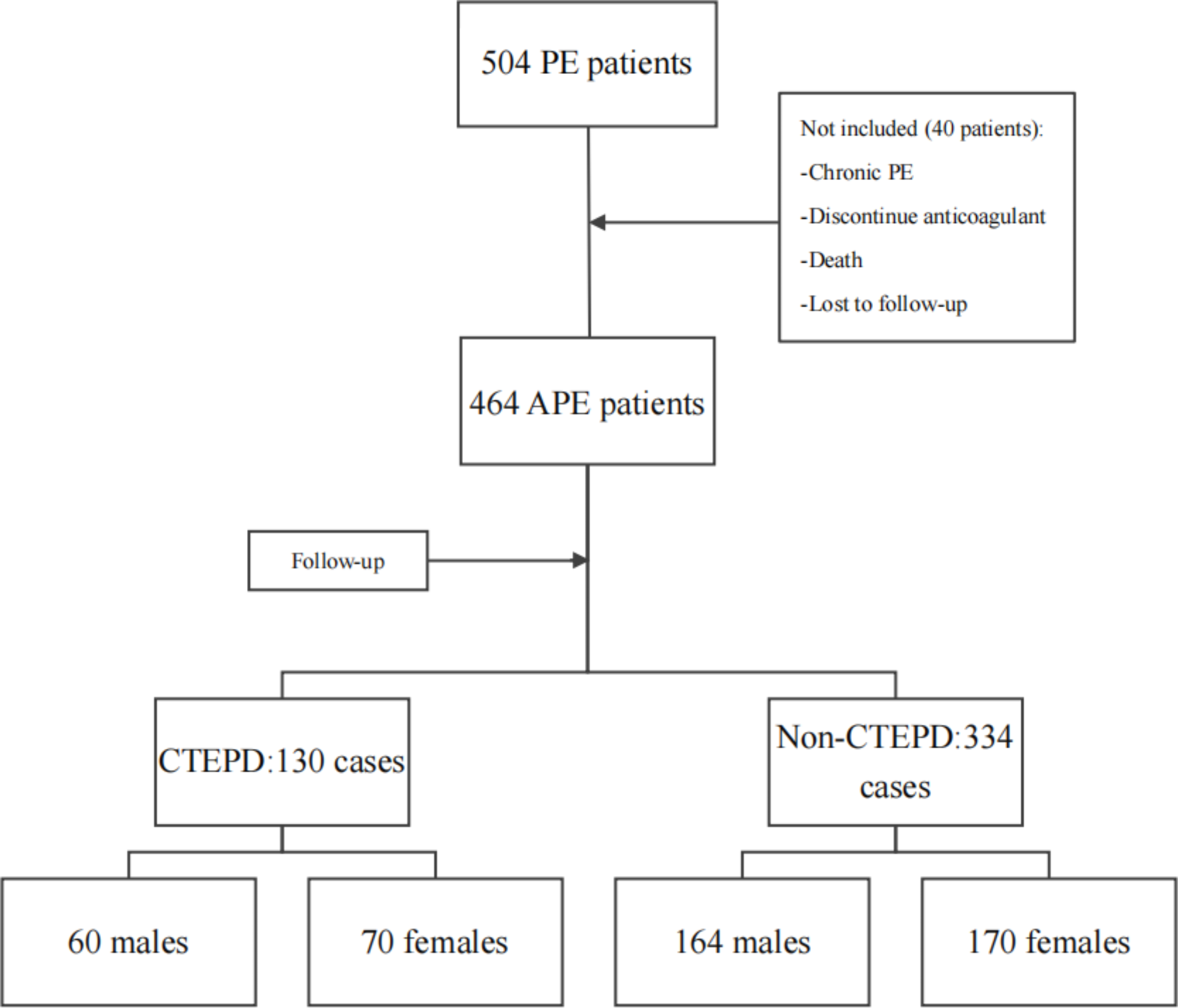
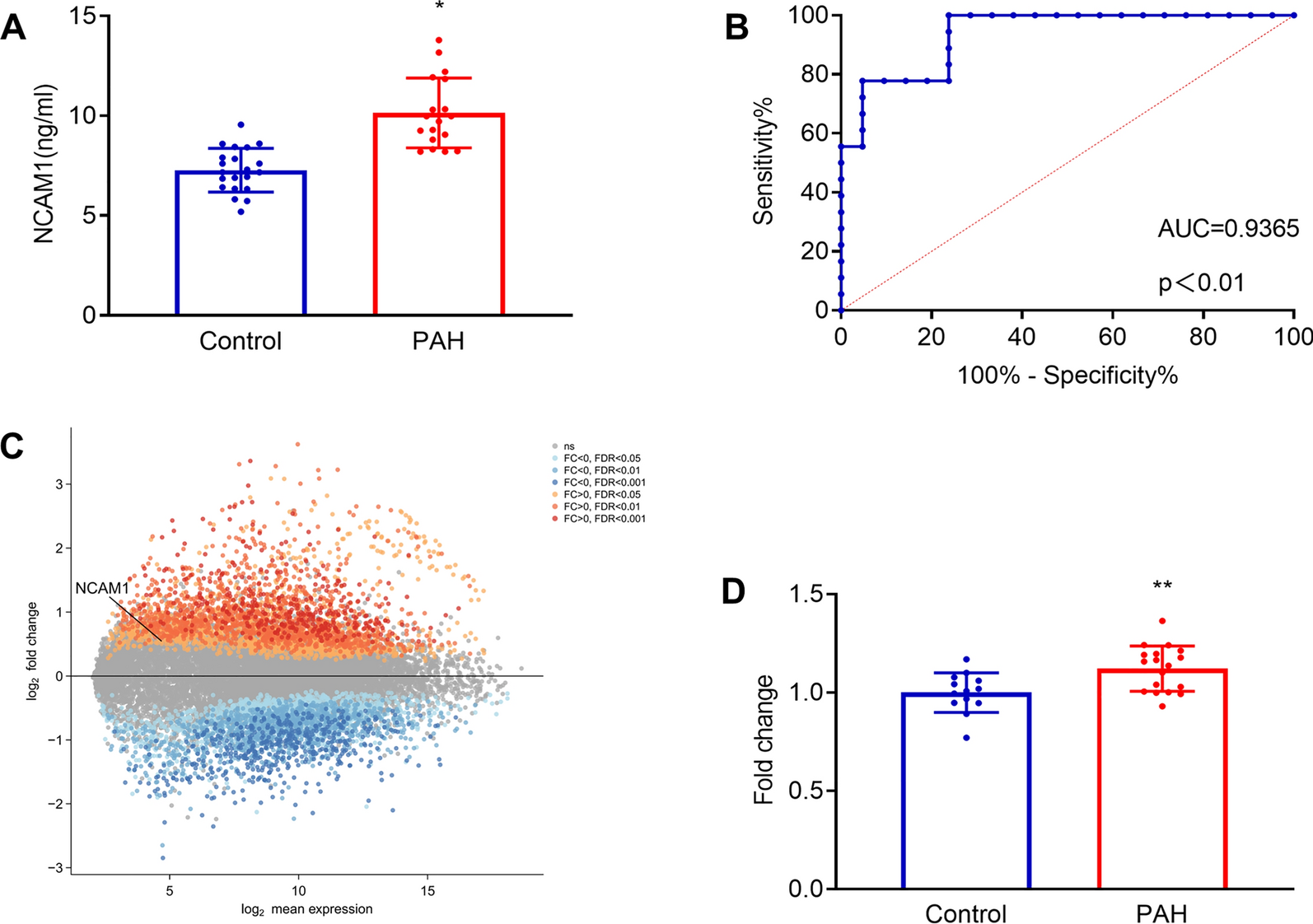
Reagents
3-Methyladenine (3-MA), compound C, and Phen green SK (PGSK) diacetate were obtained from MedChemExpress company (Shanghai, China). Selective inhibitor of ferroptosis Ferrostain-1 (Fer-1) was procured by Targetmol (Shanghai, China). Enzyme-linked immunosorbent assay (ELISA) kits for the quantification of tumor necrosis factor-alpha (TNF-α), interleukin-6 (IL-6), and interleukin-1 beta (IL-1β) were purchased from YouPin Biological Technology Co. Ltd (Shenzhen, China). Tissue Iron Content Assay Kit was purchased from Solarbio (Beijing, China). Assay kits for detecting total protein, Lyso-Tracker Green, and malondialdehyde (MDA) were purchased from Beyotime Biotech (Shanghai, China). BODIPY™ 581/591 C11 and cell counting Kit-8 (CCK8) were obtained from GLPBIO (Montclair, CA, USA). FerroOrange was obtained from Donjido (Shanghai, China). GPX4 (sc-166570, Santa Cruz Biotechnology), SLC7A11 (12509, Affinity), NCOA4 (A5695, ABclonal), FTH1 (R23306, Zenbio), AMPK (340763, Zenbio), P-AMPK (R380431, Zenbio), ULK1 (381887, Zenbio), p-ULK1 (80218-1-RR, Proteintech), β-actin (4970, Cell Signaling Technology), and GAPDH (sc-137179, Santa Cruz Biotechnology) were used as primary antibodies. Goat anti-rabbit IgG H&L (ab216773) was purchased from Abcam (Cambridge, MA, USA). The goat anti-Mouse IgG H&L (92632210) was provided by LI-COR (NE, USA). HRP-conjugated Affinipure goat anti-rabbit lgG (H + L) was obtained from Proteintech (Wuhan, Chain). Cy3-labeled goat anti-rabbit IgG (GB21303) and Cy3 Labeled Goat Anti-Mouse IgG (GB21301) were purchased from Servicebio Technology (Wuhan, China). The specific primers of SLC7A11, GPX4, and NCOA4 were synthesized and provided by Sangon Biotech (Shanghai, China).
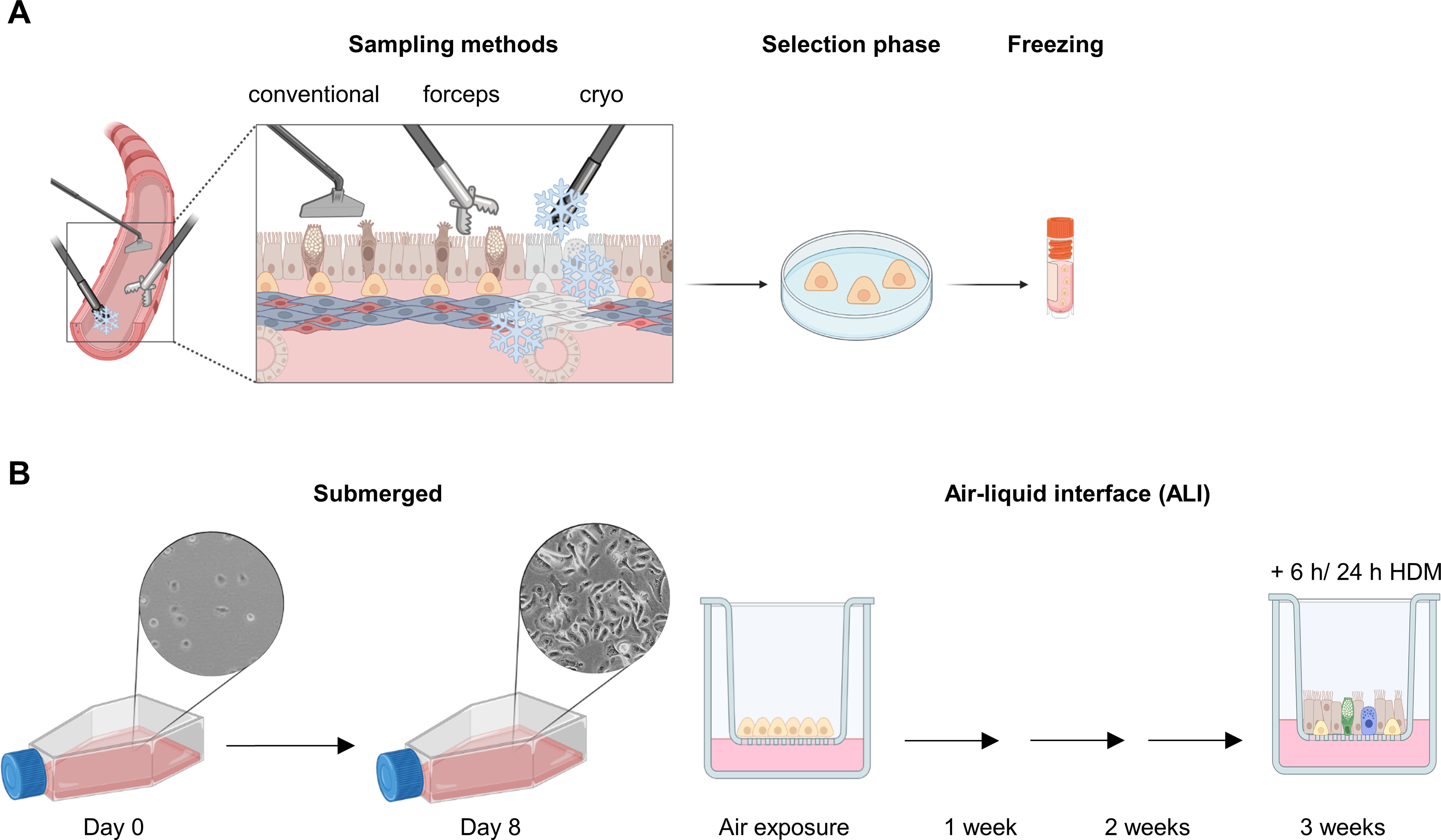
Establishment of the VILI model in mice
Male C57/BL6 mice (25 ± 2 g, 6–8 weeks old) were purchased from the Animal Center of Guangxi Medical University (Nanning, China). Mice were conditioned under controlled conditions at a room temperature of 22 °C ± 2 °C with a 12-hour light/dark cycle. They had ad libitum access to standard diet and tap water. Mice were randomly allocated into five groups: (1) control group (CON group, spontaneous breathing post-intubation, 4 h); (2) high tidal volume group (HTV group, MV with high tidal volume, 20 ml/kg, 4 h); (3) Fer-1 + HTV group [Fer-1 (1 mg/kg) was intraperitoneally injected for 14 consecutive days before MV (20 ml/kg for 4 h)] [17]; (4) 3-MA + HTV group [3-methyladenine (3-MA) (35 mg/kg) was intraperitoneally injected 1 h before MV (20 ml/kg) for 4 h), once] [28]; (5) compound C + HTV group [compound C, (20 mg/kg), intraperitoneally, once, 1 h before MV (20 ml/kg) for 4 h] [29]. The animal model of VILI was successfully established based on our previous study [30, 31]. At the end of MV or spontaneous breathing, mice were euthanized via intraperitoneal administration of a high dose of anesthetics. Subsequently, lung tissue, blood serum, and bronchoalveolar lavage fluid (BALF) samples were obtained for subsequent experimental analysis. All animal experiments were conducted with utmost care to minimize the inflammatory response.
Histopathological analysis
Hematoxylin and eosin staining were used to measure the pathological changes in the lungs of mice with VILI. The lung tissue was fixed in 4% paraformaldehyde and subsequently embedded in paraffin. Then, tissue blocks were sliced into 4 μm-thick sections and affixed onto slides. Following deparaffinization, the sections were meticulously stained with hematoxylin and eosin (H&E) before a thorough examination under a light microscope. As previously reported, the degree of lung injury was measured based on a histologic ALI scoring system [32, 33].
Immunofluorescence (IF) staining
Immunofluorescence staining was conducted as previously described [22]. Briefly, dewaxed paraffin sections were subjected to immunofluorescent staining after a sequence of deparaffinization and rehydration. The tissues were incubated at 4℃ overnight with SLC7A11 (1:200), GPX4 (1:200), NCOA4 (1:200) and FTH1 (1:200) primary antibodies. Cy3-labeled goat anti-rabbit IgG (1:400) or Cy3-labeled goat anti-mouse IgG (1:400) was used as fluorophore-conjugated secondary antibodies. The nucleus was stained with DAPI. Subsequently, the fluorescently stained lung tissue samples were examined under a fluorescent microscope (Nikon EclipseC1, Nikon).
Inflammatory responses
The wet/dry (W/D) weight ratio was calculated to crudely assess VILI lung edema. The middle lobe of the right lung was weighed (wet weight) and placed in an oven at 60 °C for 48 h (dry weight). The pulmonary permeability of mice with VILI was evaluated by measuring total protein levels in BALF supernatant using a bicinchoninic acid (BCA) assay. Cells in BLAF were counted using a hemocytometer to determine the inflammatory infiltration of VILI mice. Thereafter, the serum levels of mouse IL-1β, mouse IL-6, and mouse TNF-α were quantified using ELISA kits following the manufacturer’s instructions.
Measurement of tissue iron and malondialdehyde (MDA) concentrations
Iron and MDA concentrations in lung tissues were measured to investigate the levels of ferroptosis. Lung iron measurement was conducted using the Tissue Iron Content Assay Kit (BC4355, Solarbio, Beijing, China). MDA levels were measured using the Lipid Peroxidation MDA Assay Kit (S0131, Beyotime, Shanghai, China) following the manufacturer’s protocol.
Generation of an in vitro model of VILI
Mouse lung epithelial cells (MLE12) were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin (Invitrogen, USA). MLE12 cells were cultured with 5% CO2 at 37 °C. To stably knock-down the NCOA4 gene in MLE12 cells, we used plasmids LV2N (U6/Puro)-Ncoa4-1130-mus and LV2N (U6/Puro)-NC purchased from GenePharma (Suzhou, China) as shRNA targeting NCOA4. We employed Lipofectamine 2000 Transfection Reagent (Invitrogen, 11668030) to transduce the target plasmid and helped plasmid into 293T cells for virus production. Finally, puromycin was utilized to select positive cells. Subsequently, we successfully established an in vitro model of VILI based on our previous published study [17, 34]. Briefly, cells in the logarithmic growth phase were seeded onto six-well BioFlex plates (Flexcell International) at standard densities. Following a 24-hour incubation, cells were exposed to cyclic stretching (CS) using the FX 6000TM Tension Plus system equipped with a 25 mm loading station (Flexcell International, McKeesport, PA, United States). The stretching was applied at a rate of 30 cycles per minute (0.5 Hz), with an equal stretch and relaxation phase and a sine wave pattern. In our study, the pathological CS was conducted at 20% changes in basement membrane surface area corresponding to 80% of total lung capacity. Cells were stretched for 4 h at 37˚C in a humidified incubator containing 5% CO2. The entire experimental procedure was meticulously controlled by computer software. In addition, the culture medium was supplemented with 10 µmol/L of compound C to inhibit AMPK [29]. Culture supernatants and cells were obtained after 4 h of cell stretching treatment.
Cell viability assay
Cell viability was determined using the CCK-8 assay kit. After treatment, cells were seeded at a density of 1 × 104 cells per well onto 96-well plates for 12 h. Subsequently, the CCK-8 solution was added directly to the culture medium and incubated at 37 °C for 2 h. Finally, using a microplate reader, the optical density values were detected at 450 nm.
Transmission electron microscopy (TEM)
TEM assays were conducted as previously described [17]. Lung tissues were collected after modeling and then cut into 1–2 mm cubes, fixed in 2.5% glutaraldehyde and 2.5% paraformaldehyde in 0.1 M sodium cacodylate buffer (pH 7.4) for at least 2 h at 4 ℃. Next, cubes were put into the same buffer with 2% uranyl acetate for 2 h at room temperature before dehydration in graded alcohols and propylene oxide. After embedding in resin, the samples were cut into ultrathin slices, stained with uranyl acetate and observed using a HT7800 transmission electron microscope (Hitachi, Japan). Similarly, MLE12 cells were harvested for TEM analysis to evaluate cellular damage and the detailed structure of autophagosomes.
Measurement of intracellular iron
Intracellular iron levels were measured using Phen Green SK (PGSK) [35]. MEL12 cells were seeded at onto plates after pretreatment, washed with PBS, and dark incubated with 20 µM PGSK for 20 min at 37 ℃. The results were visualized using fluorescence microscopy (Leica, Germany).
Lipid peroxidation assay
Suitably treated cells were incubated with 10 µM BODIPY™ 581/591C11 fluorescence probe for 30 min in a humidified incubator (37 ℃, 5% CO2). Nuclear staining was conducted with DAPI, followed by washing with PBS. Finally, the results were visualized using fluorescence microscopy (Leica, Germany).
Localization of intracellular and lysosomal Fe2+
Suitably treated cells were co-stained with FerroOrange (1 µM) and Lyso-Tracker Green (100 nM) in a serum-free medium. They were incubated in an incubator with 5% CO2 at 37 °C for 30 min. Cells were washed with PBS. The results were immediately observed using fluorescence microscopy (Leica, Germany).
Real-time PCR
The mRNA levels of NCOA4, SLC7A11, and GPX4 were determined using real-time PCR with specific primers under Real-Time PCR System as detailed in previously. The primer sequences were as follows:
SLC7A11, forward: 5′-ATGGTCAGAAAGCCAGTTGTG-3′, reverse: 5′-GCTCCAGGGCGTATTACGAG-3′;
GPX4, forward: 5′-CTCCGAGTTCCTGGGCTTGTG-3′, reverse: 5′- CCGTCGATGTCCTTGGCTGAG-3′;
NCOA4, forward: 5′-AGTTCCTTGTCAGAGTGGCTTATGG-3′, reverse: 5′-ACCCAGTCGGCAGTGTTAAAGG-3′;
β-actin, forward: 5′-CCACGACAAGGAGCTGCTTCTG-3′, reverse: 5′-ACCCTGTCCGCCATCACATCA-3′;
GAPDH, forward: 5′-CCTTCATTGACCTCAACTACATGG-3′, reverse: 5′-CTCGCTCCTGGAAGATGGTG-3′;
β-actin and GAPDH were used as internal controls. We employed the 2−△△Ct method for quantification.
Western blotting
Total proteins were extracted from the lung tissue and MLE12 cells using radio-immunoprecipitation assay (RIPA) buffer supplemented with protease inhibitor and protein concentrations were assessed using BCA protein assay kit. Then, equal amounts of protein and the molecular weight marker were added to the lanes of sodium dodecyl sulfate (SDS)-polyacrylamide gel. They were subsequently transferred onto polyvinylidene fluoride (PVDF) membranes. After being blocked with Western blotting rapid blocking buffer for 30 min at room temperature, the membranes were incubated by SLC7A11 (1:700), GPX4 (1:200), NCOA4 (1:1000), FTH1 (1:1000), AMPK (1:500), P-AMPK (1:500), ULK1 (1:500), P-ULK1 (1:1000), and β-actin (1:1000), and GAPDH (1:1000) primary antibodies. Next, they were incubated with the following secondary antibodies: goat anti-rabbit IgG H&L (1:15000) or goat anti-Mouse IgG H&L (1:20000). Finally, Western blotting bands were visualized using an Odyssey two-color infrared laser imaging system (LICOR, USA).
Statistical analyses
SPSS 26.0 software (IBM, USA) was used for statistical analysis. All data are presented as mean ± standard deviation (SD). Differences were analyzed using Analysis of Variance (ANOVA) followed by the LSD-t test and the SNK test for pair-wise comparisons. P value less than 0.05 was considered statistically significant.



留言 (0)