
記住我






This prospective, double-blind, single-center, randomized controlled trial will be conducted at the Liver Transplantation Center of West China Hospital, Sichuan University. The aim of this study is to compare the fluorescence navigation effects of our center’s novel ICG-HSA administration scheme with the current guideline-recommended scheme for NIF-negative staining in LALR [3] as well as to analyze the safety and efficacy of this new administration scheme.
Study populationInclusion criteriaPatients must meet all of the following criteria to be eligible for inclusion in this study: (1) age of 18 to 75 years with a malignant liver tumor requiring LALR, (2) preoperative Child–Pugh class A or B liver disease, (3) no contraindication to laparoscopic liver resection, (4) expected survival of ≥ 3 months, (5) Eastern Cooperative Oncology Group performance status score of 0 or 1, and (6) laboratory testing within 7 days prior to enrollment meeting the following conditions: white blood cell count of ≥ 2.5 × 109/L, absolute neutrophil count of ≥ 1.5 × 109/L, platelet count of ≥ 75 × 109/L, hemoglobin level of ≥ 90 g/L, international normalized ratio of ≤ 1.5 × upper limit of normal (ULN), serum creatinine level of ≤ 1.5 × ULN, and total bilirubin level of ≤ 1.5 × ULN.
Exclusion criteriaPatients who meet any of the following criteria will be excluded from participation in this study: (1) no obvious ischemic demarcation line after intraoperative blocking or isolation of the target liver pedicle, or liver fluorescence reaching an intensity that interferes with the operation before intraoperative intravenous injection of ICG; (2) ICG retention rate at 15 min of ≥ 20%; (3) severe cardiopulmonary disease precluding surgery under general anesthesia; (4) clinically significant ascites or pleural effusion; (5) active bleeding or coagulation dysfunction; (6) hepatic encephalopathy; (7) allergy to ICG; (8) history of gastrointestinal bleeding within the past 6 months or a tendency for gastrointestinal bleeding; (9) severe gastroesophageal varices requiring treatment; (10) objective evidence of past or present severely impaired lung function; and (11) any clinical or laboratory abnormalities that may affect the safety evaluation.
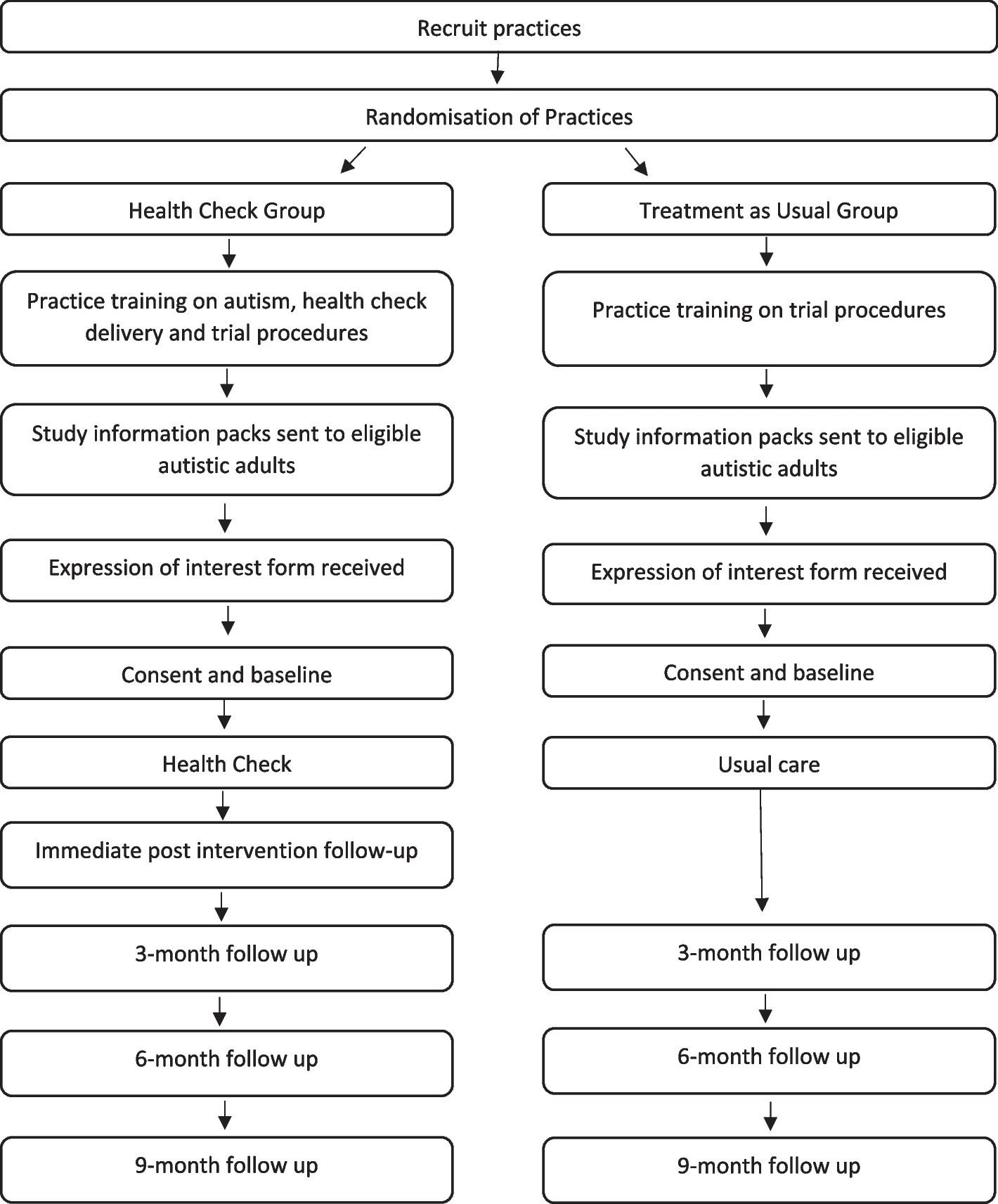
Study drugsFor this study, we have developed a novel ICG-HSA complex for use in fluorescence image-guided LALR. The clinical formulation of the ICG-HSA complex was designed based on the results of in vitro validation experiments. The complex was prepared using commercially available HSA (10 g/50 mL) and ICG (25 mg/10 mL). Given that the molar mass of HSA is approximately 66,500 g/mol [15] and the molar mass of ICG is 774.96 g/mol [10], we calculated the required volumes of each component to achieve a molar ratio of ICG:HSA = 1:6. This ratio was determined to be optimal for clinical use. To prepare the clinical formulation, 2 mL of the commercial HSA injection and 0.3 mL of the commercial ICG solution were mixed together. The mixture was then diluted with 15 mL of normal saline to maintain the desired molar ratio while providing a sufficient volume for clinical administration. The final ICG-HSA complex solution was stored in a 20-mL syringe (Fig. 1), which was connected to an intravenous infusion pump to ensure uniform injection during the surgical procedure.
Fig. 1
Configuration diagram of ICG-HSA complex. A The raw materials used to create ICG-HSA are readily available: HSA injection (10 g/50 mL) and ICG injection (25 mg/10 m). The molar ratio of ICG:HSA = 1:6; therefore, the required volume of the finished HSA injection is approximately 0.02 mL, and the finished ICG injection is approximately 0.031 mL. B HSA injection (2 mL) is thoroughly mixed with ICG injection (0.3 mL) at room temperature. C The ICG-HSA complexes are diluted with 15 mL of normal saline to form the final study drug
RandomizationThis study will employ a customized electronic network central randomization system (https://study.empoweredc.com/) for block randomization allocation using a block length of 4. Patients will be randomly assigned in a 1:1 ratio to either the treatment group (study medication group) or the control group (guideline medication group). After enrolling patients who meet the inclusion criteria, the surgical team will complete the target hepatic pedicle occlusion operation. A dedicated research nurse will then perform the registration and randomization in the randomization system according to the order of trial entry (note: this must be performed according to the order of enrollment because the first to enroll may not be the first to undergo surgery). The system will automatically assign the corresponding random number and group to the patient.
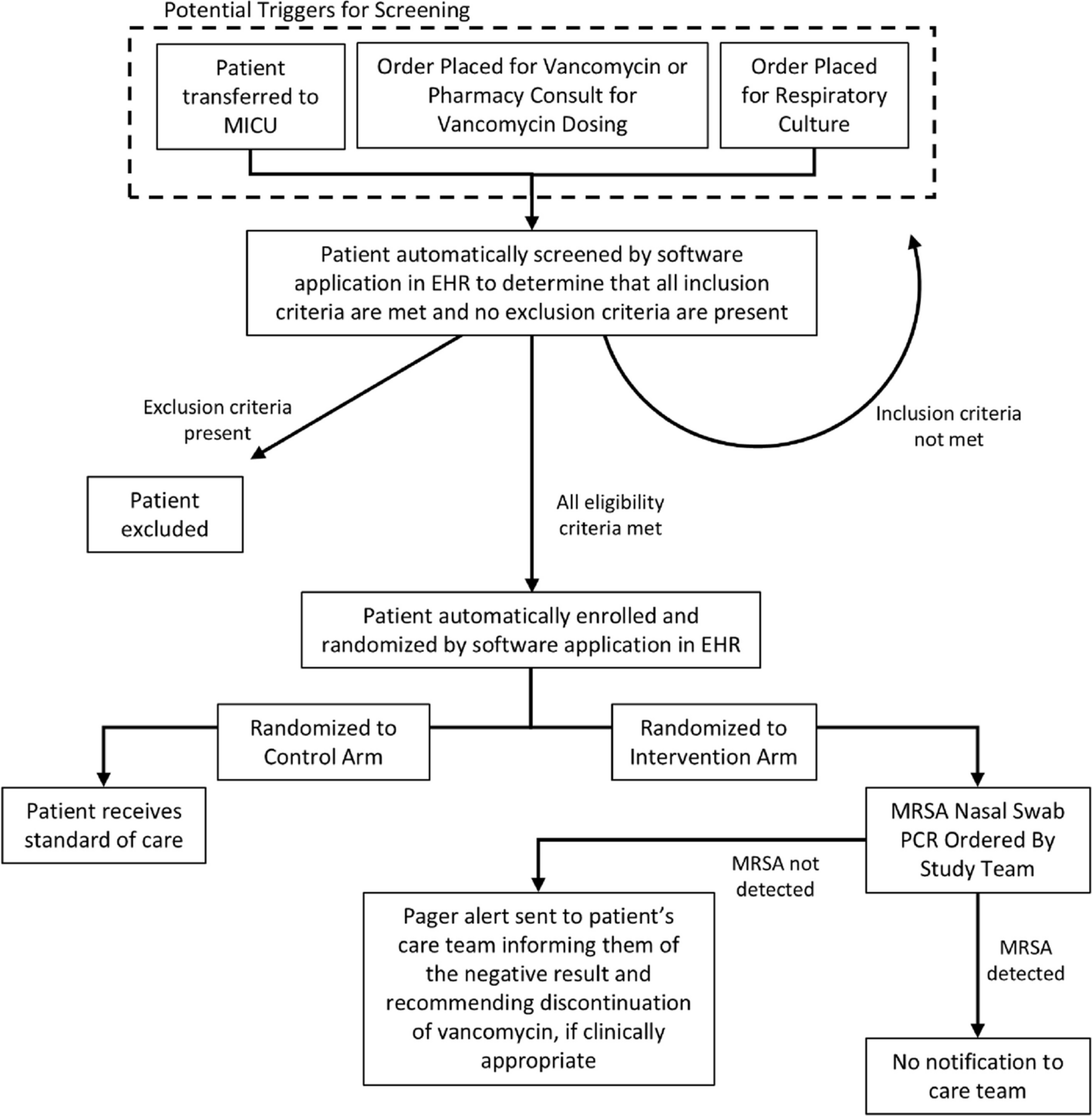
BlindingThis study will adopt a double-blind design for both the surgical team (outcome assessors) and the patients. The random group assignments will be visible only to the dedicated research nurse, who will provide the different ICG administration schemes for the two groups based on the random numbers and group assignments during the surgery. An opaque curtain will be used to cover the anesthesia operation area at the patient’s head side, thereby blinding the surgical operators to the random group assignments. The entire process of grouping and administration will be visible only to the dedicated research nurse (Fig. 2).
Fig. 2
Diagram of the blinding method. A An opaque curtain is used to cover the anesthesia operation area at the patient’s head side, preventing the surgical team from knowing the group assignment. The entire process of grouping and administration is visible only to the dedicated randomization nurse. B The nurse uses an infusion pump to administer the ICG-HSA complex solution through the patient’s central venous catheter
Concealment of group assignmentsTo reduce assessment bias, the outcome assessors and data collectors will be blinded such that they are unaware of the patients’ group assignments, thereby minimizing the impact of bias on the results. The primary endpoint will be determined by three independent experts in the field of fluorescence-guided laparoscopic liver resection, each of whom will independently evaluate and score the fluorescence imaging results in the surgical video recordings.
Unblinding principlesEmergency unblinding may only be performed if the patient develops an adverse event or if the investigator deems it necessary to clarify the patient’s specific treatment group for further management of the situation. The investigator can log into the randomization system and unblind the patient by entering the password. The unblinding page must document the reason for emergency unblinding, the time, and the personnel involved in the unblinding process.
Criteria for terminating the study (1)Serious adverse event: If a patient develops a serious adverse event during the study that significantly impacts their health and may be related to the study intervention, it may be necessary to consider terminating the study.
(2)Lack of efficacy: If, after a certain period of observation and treatment, the study intervention has failed to achieve the expected efficacy or no significant differences have been observed compared with the control group, it may be necessary to consider terminating the study.
(3)Exceeding predetermined safety limits: If the frequency or severity of adverse events caused by the study intervention exceeds the predetermined safety limits, the study may need to be terminated to protect the safety of the patients.
(4)Poor compliance: If patients are unable to follow the study protocol requirements or lack necessary cooperation or compliance during the study, and this significantly impacts the reliability of the data, it may be necessary to consider terminating the study.
The criteria for terminating the study should be established under the guidance of the study protocol and the ethics review board, and these criteria should be clearly documented in the study protocol in advance. Upon terminating the study, relevant institutions and participants should be promptly notified, and necessary data analysis and interpretation should be conducted.
Principles for handling dropouts (1)Recording and statistics: For each patient who drops out, the reason for dropout will be accurately and promptly recorded, and a statistical analysis will be conducted. This will help to understand the overall situation of dropouts as well as the proportions and trends of different reasons for dropping out.
(2)Adverse events: For dropouts caused by serious adverse events, the severity and relevance of the event will be carefully evaluated, and consideration will be given to whether the trial needs to be halted or the protocol adjusted. This will require comprehensive consideration of patient safety and the scientific validity of the trial.
(3)Voluntary withdrawal: For patients who voluntarily withdraw from the trial, consultations will be conducted as early as possible to understand their reasons for withdrawal, and these reasons will be recorded. This will help to analyze the patients’ acceptance of the trial intervention and identify potential areas for improvement.
(4)Exclusion criteria: Patients who meet the exclusion criteria will be screened and excluded before the trial begins. This will prevent patients who do not meet the trial requirements from interfering with the trial results.
Trial interventionsBased on preoperative three-dimensional reconstruction imaging, an anatomical liver resection plan will be formulated to resect the liver segment or lobe containing the tumor. All surgeries will be performed by the same hepatobiliary surgical team. The surgeons that will be involved in this study have more than 10 years of experience and have each completed more than 100 laparoscopic hepatectomies. After induction of general endotracheal anesthesia, the patients will be placed in the supine position for laparoscopic hepatectomy. The intra-abdominal pressure will be set at 10 to 14 mmHg. Central venous pressure will be monitored and maintained below 5 cmH2O. An ultrasonic scalpel (Harmonic Scalpel; Ethicon, Cincinnati, OH, USA) will be used to adequately mobilize the hepatic ligaments. We will use the guideline-recommended “negative staining method” to complete the LALR [3]. The hepatic pedicle of the lobe or segment planned for resection will be isolated and occluded using the Glissonean pedicle transection technique after confirming the diseased area.
After completing the above steps, patients assigned to the control group will receive a peripheral intravenous injection of 1 mL ICG solution (2.5 mg/mL) administered by a dedicated nurse, followed by observation of liver fluorescence imaging. Patients assigned to the trial group will receive a steady peripheral intravenous infusion of the prepared novel ICG-HSA complex at a rate of 1 mL/min using an intravenous infusion pump, administered by a dedicated nurse. The infusion will be stopped when fluorescence imaging appears on the liver surface under the fluorescence laparoscopic system (FloNavi® 214 K; OptoMedic Technologies, Foshan, Guangdong, China) (Fig. 3). Intraoperative ultrasound will be used to confirm whether the tumor is located in the liver segment of fluorescence imaging.
Fig. 3
Schematic of a fluorescence “negative staining” laparoscopic anatomical left hemihepatectomy
Once it has been confirmed that the tumor is located in the non-fluorescent area and that the resection margin is sufficient, the liver segment will be resected along the fluorescence boundary under full liver blood flow occlusion using the Pringle maneuver. After completing the liver resection, the liver specimen will be extracted through the umbilical incision. The pneumoperitoneum will then be deflated, puncture sites sutured, and operation completed.
Primary endpointThe primary outcome of this trial will be the effectiveness of intraoperative fluorescence imaging. After the patient is discharged, the research team will provide the complete unedited surgical video to three experts in the field of fluorescence-guided laparoscopic liver resection. These experts will score the video according to our established scoring criteria (Fig. 4), and the average score from the three experts will be used as the primary outcome for the patient.
Fig. 4
Expert scoring criteria for fluorescence imaging effectiveness. Based on a maximum score of 6 points, the experts will use the complete unedited surgical video to make corresponding deductions or additions according to the scoring standards shown in the figure
Secondary endpointsThe secondary endpoints will be conversion to open surgery, total operative time, intraoperative blood loss, intraoperative transfusion, tumor resection margins, post-hepatectomy liver failure [16], length of postoperative hospital stay, incidence of complications (postoperative complications will be graded based on severity according to the Clavien–Dindo classification [17]), unplanned reoperation rate, postoperative mortality rate, overall survival time, and disease-free survival time.
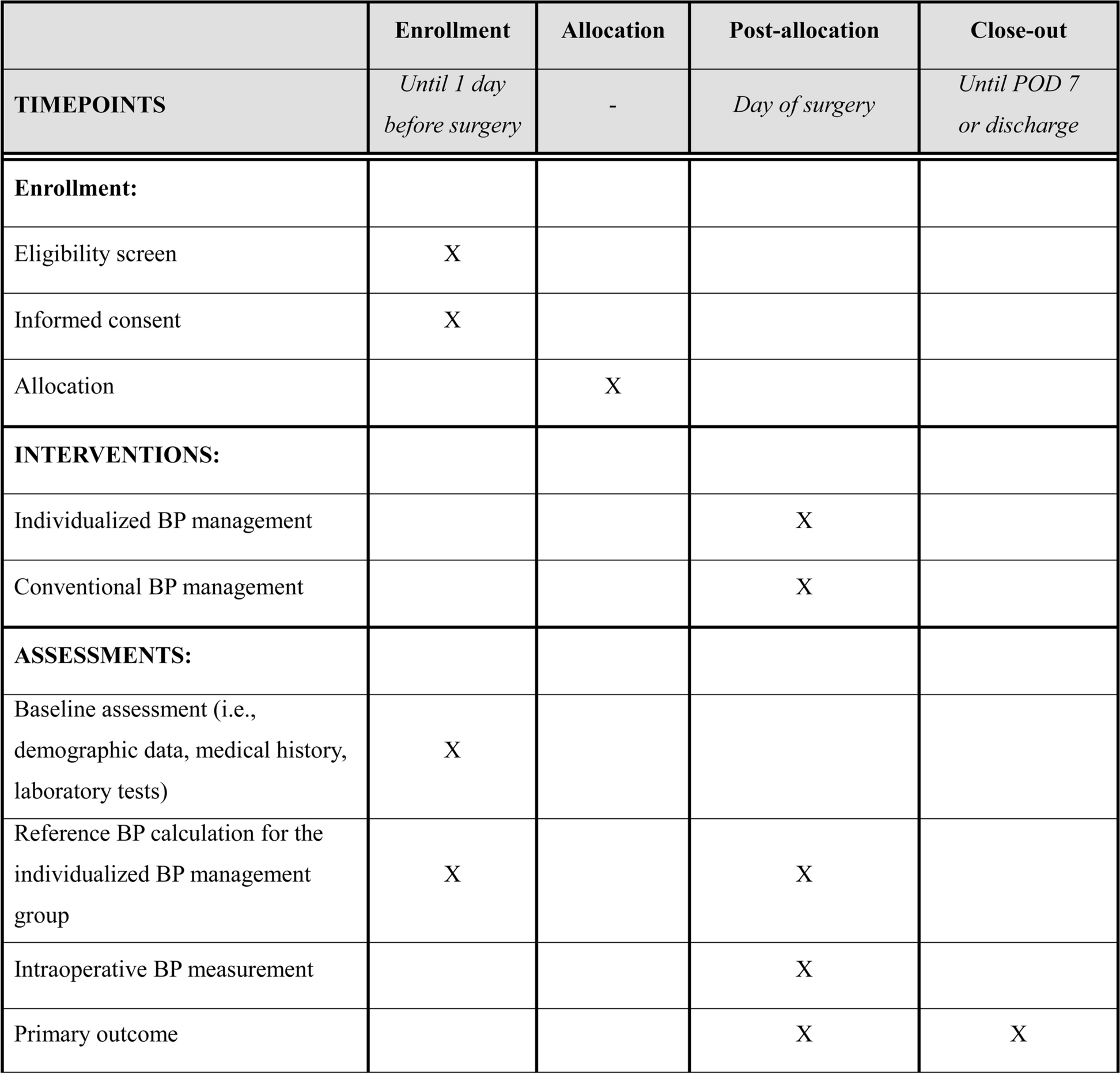
Data collection and follow-upData collection will be performed prospectively for all patients, encompassing their medical history, physical examination findings, laboratory results, pathological assessments, perioperative clinical details, and any complications. Information will be recorded on paper datasheets and stored securely. The patients will undergo evaluations at the time of inclusion, at 1 week postoperatively, and at 3, 6, 12, 24, and 36 months postoperatively. Follow-up by telephone or outpatient visits will occur at 1 month postoperatively and then at 3, 6, 12, 24, and 36 months postoperatively. Throughout follow-up, liver function tests and abdominal ultrasound examinations will be conducted for a duration of up to 3 years or until the patient’s death. The schedule of enrolment, interventions, and assessments for this trial is summarized in the SPIRIT figure below (Fig. 5). This figure provides an overview of the overall timeline and key activities at each visit, from initial eligibility screening through to study close-out.
Fig. 5
SPIRIT schedule of enrolment, interventions, and assessments. − t1: screening period (14 days before surgery); 0: surgery day; t1: 1 week postoperatively; t2: 1 month postoperatively; t3: 3 months postoperatively; t4: 6 months postoperatively; t5: 12 months postoperatively; t6: 24 months postoperatively; t7: 36 months postoperatively;1Pulse, blood pressure, body temperature. 2Routine blood tests (WBC, RBC, HGB, PLT, NL, LY) and urine routine tests (LEU, BLD, GLU, PRO). 3Blood biochemistry (ALT, AST, GGT, TBIL, ALP, Urea, Cr, GLU). 4Only for women of childbearing age. 5ICG administration protocol (experimental group, control group), conversion to open surgery, liver parenchyma disconnection time, total operation time, intraoperative bleeding volume, specimen cutting margin (malignant tumor). 6Complications (incidence, Clavien–Dindo classification [17], treatment measures), postoperative mortality, postoperative liver function recovery
The measurement time points and methods for various short-term and long-term secondary endpoints are detailed in Table 1. All clinical data, including patient demographics, surgical details, tumor characteristics, liver tumor clinical staging, perioperative observation indicators, and results from short-term and long-term follow-up assessments, will be entered and collected using an internet-based electronic data capture (EDC) system (https://study.empoweredc.com/) and centralized at the principal investigator’s center. The principal investigator and co-investigators, with assistance from the research assistants, will conduct continuous clinical data monitoring as well as interim and final analyses.
Table 1 Measurement times and methods of short- and long-term secondary indicatorsStatistical analysesSample size calculationThe sample size has been calculated based on the primary outcome of intraoperative fluorescence imaging effect scores. Assuming a mean score of 4 in the control group and an expected mean score of 4.5 in the experimental group, and with a common standard deviation of 0.8, a sample size of 64 patients per group would provide 80% power to detect a statistically significant difference between groups using a two-sided t-test at a significance level of 0.05 (NCSS and PASS 15 statistical software; NCSS, LLC, Kaysville, UT, USA). Allowing for a 10% dropout rate, 142 patients (71 per group) will be recruited.
Statistical methodsStatistical analysis will be performed using SPSS 26.0 software (IBM Corp., Armonk, NY, USA). Continuous variables will be expressed as mean ± standard deviation or median (interquartile range) and compared using the independent-samples t-test or Mann–Whitney U test. Categorical variables will be expressed as number (percentage) and compared using the chi-square test or Fisher’s exact test. The primary outcome of intraoperative fluorescence imaging scores will be compared between groups using the independent-sample t-test. Secondary short-term outcomes such as conversion to open surgery, total operation time, intraoperative blood loss, tumor margins, postoperative liver failure, length of hospital stay, complication rate, unplanned reoperation rate, and mortality will be compared between groups using appropriate statistical tests based on the type and distribution of the data. Long-term outcomes of overall survival and disease-free survival will be analyzed using the Kaplan–Meier method, and differences between groups will be assessed using the log-rank test. Multivariable Cox proportional hazards models will be used to identify independent prognostic factors associated with overall and disease-free survival. All statistical tests will be two-sided, and p-values of < 0.05 will be considered statistically significant. Intention-to-treat analysis will be used, with all randomized patients included in the analysis according to their assigned group. Per-protocol analysis will also be performed as a sensitivity analysis. Subgroup analyses based on factors such as the type of liver resection may be conducted if deemed appropriate.
An interim analysis will be performed to assess safety and efficacy when 50% of the planned sample size has been enrolled. The trial may be stopped early if there are significant differences in safety outcomes between groups or if the efficacy boundary is crossed. The final analysis will take place once complete survival data have been collected after the 3-year follow-up of the last enrolled patient.
Quality control of study dataThe principal investigator and co-investigators, with the assistance of the research assistants, will conduct continuous clinical data monitoring as well as interim and final analyses using the EDC system (https://study.empoweredc.com/). The researchers will complete and submit the online version of the case report form promptly after each patient’s visit. The researchers must respond to queries from monitors, data managers, and medical reviewers in a timely manner. After data cleaning has been completed, the researchers will sign off to confirm the data for each patient.
Data review and database lockUpon completion of the clinical trial, the study lead, statisticians, and data managers will jointly conduct a pre-statistical analysis data review. The key aspects will be determining the analysis dataset for each case (including the full analysis set, per-protocol set, and safety set), assessing missing values, and handling outliers. Once the data review confirms that the database is accurate, the database will be locked. After locking, the database cannot be modified arbitrarily, and any modification decisions must be documented. The locked database will be properly preserved for future reference and handed over to statisticians for analysis.
Data safety monitoringA data safety monitoring plan will be developed based on the level of risk. All adverse events will be recorded in detail, appropriately managed, and tracked until resolved or stabilized. Serious adverse events and unexpected events will be reported promptly to the ethics review board, health authorities, and drug regulatory agencies as required. The principal investigator will periodically conduct cumulative reviews of all adverse events and, if necessary, convene investigator meetings to assess the risk and benefits of the study. In double-blind trials, emergency unblinding may be performed if necessary to ensure the safety and rights of the patients.
Ethics and disseminationThe study will be conducted in accordance with the ethical principles set out in the Declaration of Helsinki and will be consistent with the International Council for Harmonisation/Good Clinical Practice as well as the regulatory requirements for participant data protection. It will be conducted in West China Hospital, which has been approved by the Ethics Committee of West China Hospital, Sichuan University. All participants will receive the information necessary to provide informed consent, including key details about the clinical trial such as financial costs, the operative procedure, benefits, and risks. They will be entitled to withdraw from the study at any time without providing a reason. We will share the trial results with the public within 1 year after completion of the clinical trial and publish the results in a peer-reviewed journal. However, systematic individual patient data sharing will not be performed.
留言 (0)