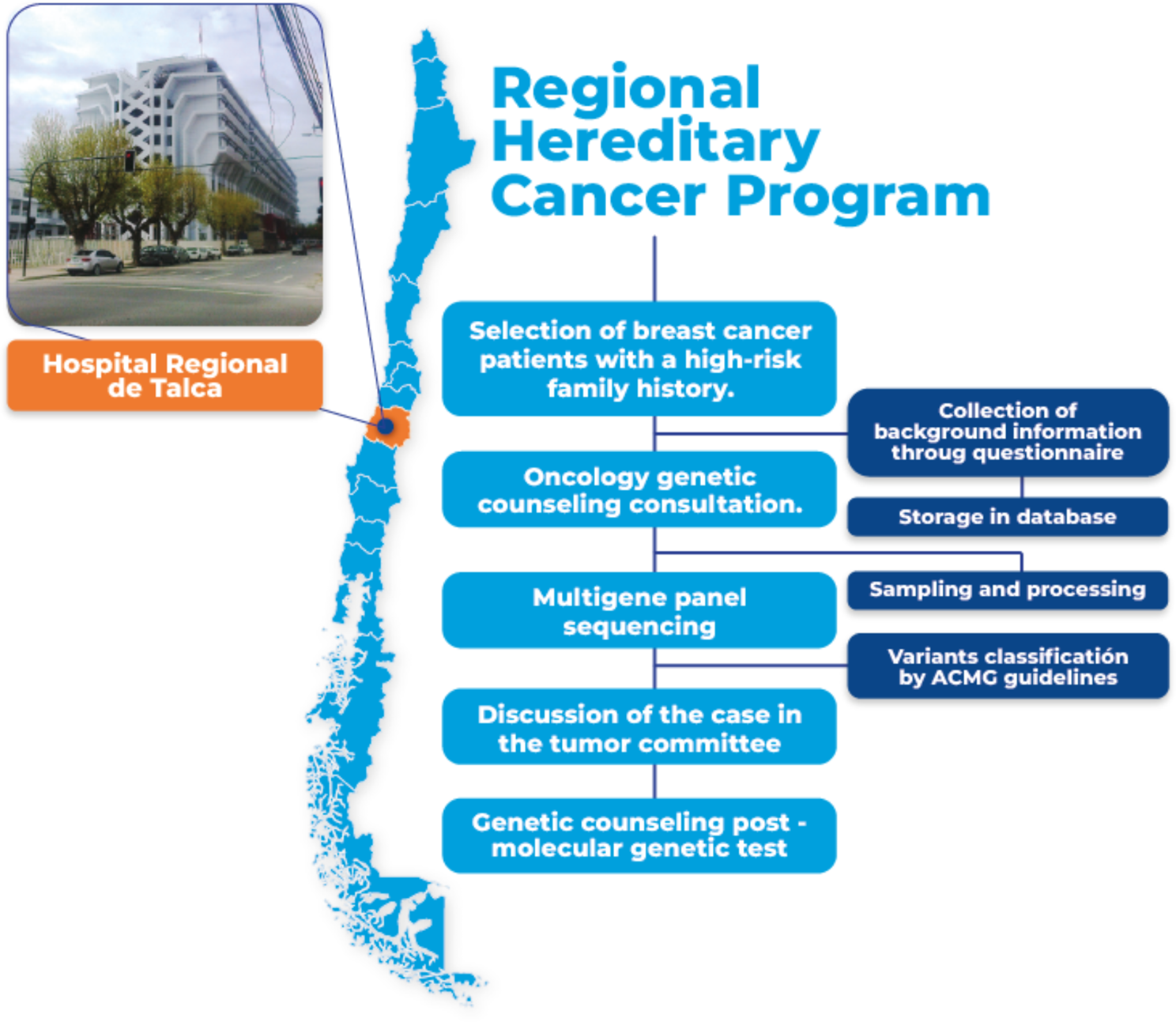
Breast cancer population study
Patients with suspected hereditary BC syndrome were recruited from the Hospital Regional Talca, which cares for cancer patients from the entire Maule Region. The patients were referred for oncology counseling by telemedicine and molecular genetics study (analysis of the multigene panel of 162 genes associated with hereditary cancer). In the counseling consultation, clinical, pathological and family information was obtained with which the patient’s genealogy was generated, incorporating healthy and cancer patients in the family. This research was approved by the institutional ethics committee of Universidad Católica del Maule (176/2021). All participants gave their written informed consent and completed a questionnaire related to their medical and reproductive history, ethnicity, and risk factors. In total, 48 cases with BC were selected for this pilot study.
Syndrome-specific indications were included such as: (i) families with BC and ovarian cancer, tubal carcinoma, or primary peritoneal carcinoma regardless of the age of onset, (ii) families with BC, endometrial cancer, and/or thyroid carcinoma, (iii) families with BC and pediatric tumors (such as sarcomas, leukemias and central nervous system tumors), and (iv) families with BC and gastrointestinal disease (such as hamartomatous polyps, diffuse gastric cancer, colon cancer).
Sampling and DNA isolation
The blood sample was extracted into a tube with EDTA anticoagulant and taken to the Biomedical Research Laboratories of the Universidad Católica del Maule for processing it. Genomic DNA was extracted from peripheral blood leukocytes using E.N.Z.A Blood DNA Maxi Kit, Omega Bio-Tek (D2492-03) according to the manufacturer’s instructions and was quantified using NanoDrop™ 2000.
Genes analyzed and library preparation by NGS sequencing
The sequencing was made in Sistemas Genómicos - Spain. The 162 genes OncoClever-GeneSGKit library was used. For the library preparation ‘Paired end’ reads 101 nt length were generated. Then, targeted regions were enriched using targeted sequencing protocol and sequenced in an Illumina NextSeq sequencing platform. Reads were aligned against the human reference genome version GRCh38/hg38. Read alignment was performed using BWA and ‘in-house’ scripts.
Genes included in the library were: ACD, BMPR1A,, DDB2, ERCC4, FANCM, HNF1B, MET, NF2, PMS2CL, RAD51C, SDHAF2, TERC, WRN, AIP, BRCA1, DICER1, ERCC5, FH, HOXB13, MITF, NFIX, POLD1, RAD51D, SDHB, TERT, WT1, AKT1, BRCA2, DIS3L2, ERCC6, FLCN, HRAS, MLH1, NHP2, POLE, RB1, SDHC, TGFBR2, XPA, ALK, BRIP1, DKC1, FAN1, G6PC3, IDH1, MLH3, NOP10, POLH, RECQL, SDHD, TINF2, XPC, APC, BUB1, DLST, FANCA, GALNT12, JAGN1, MNX1, NSD1, POT1, RECQL4, SLC25A11, TMEM127, XRCC2, AR, CDC73, EGLN1, FANCB, GCM2, KIF1B, MRE11, NTHL1, PRCC, RET, SLX4, TP53, XRCC3, ATM, CDH1, EGLN2, FANCC, GDNF, KIT, MSH2, PALB2, PRKAR1A, RFWD3, SMAD4, TSC1, ATR, CDK4, ELANE, FANCD2, GFI1, LZTR1, MSH3, PARN, PRSS1, RNF139, SMARCB1, TSC2, ATRX, CDKN1B, EPAS1, FANCE, GPC3, MAD2L2, MSH6, PDGFRA, PTCH1, RNF43, SMARCA4, UBE2T, AXIN2, CDKN2A, EPCAM, FANCF, GPR101, MAX, MSR1, PHOX2B, PTCH2, RPS20, SMARCE1, VHL, BAP1, CHEK2, ERCC1, FANCG, GREM1, MC1R, MUTYH, PIK3CA, PTEN, RTEL1, SRP54, VPS45, BARD1, CSF3R, ERCC2, FANCI, HAX1, MDH2, NBN, PMS1, RAD50, SCG5, STK11, WAS, BLM, CTNNA1, ERCC3, FANCL, HNF1A, MEN1, NF1, PMS2, RAD51, SDHA, SUFU, and WRAP53.
Interpretation of identified variants
Germline variants with allele frequency < 0.02 based on allele frequencies and coverage > 20 reads were selected. The variants found were classified according to the American College of Medical Genetics and Genomics (ACMG) guidelines as pathogenic, likely pathogenic, benign, likely benign or variants of unknown significance [19]. Variants were described according to the nomenclature recommendations of the Human Genome Variation Society [20].
Validation and segregation analysis by Sanger sequencing
To validate the clinically relevant variants (pathogenic and likely pathogenic) Sanger sequencing was performed. In addition, the identified pathogenic variants were studied by Sanger in first-degree relatives. The primers used for sequencing are detailed in Table 1.
Table 1 List of PCR primers used to validate mutations by Sanger sequencingDetection of large genomic rearrangement using copy number variation analysis
The algorithm for the detection of copy number variants (CNVs) using NGS (multigene panel) is based on the comparison of the reading depth of a certain region of the genome of the sample under study in comparison with a set of samples used as a reference [21]. The underlying hypothesis of this method is that the read depth of a genomic region is correlated with the number of copies of that region. CNVs were evaluated for the genes ATM, BARD1, BLM, BRCA1, BRCA2, BRIP1, CDH1, CHEK2, EPCAM, ERCC4, FANCM, MLH1, MSH2, MSH6, NBN,NF1, PALB2, PMS2, PTEN, RAD50, RAD51, RAD51C, RAD51D, RECQL, STK11, TP53, XRCC2, and XRCC3.



留言 (0)