
記住我


Forty-eight male C57BL/6 mice (6 weeks old, weighing 18–22 g) were acquired from Liaoning Changsheng Biotechnology Co., Ltd. (Benxi, Liaoning Province, China) and were randomly allocated into four groups: control, model, bexarotene, and model + bexarotene (Fig. 1A). The COPD mouse model was generated according to a previous study with some modifications [11, 12]. In the model and model + bexarotene groups, the mice were subjected to passive exposure to mainstream CS for 30 min twice daily, with an interval of 6–8 h, for 30 consecutive days. This exposure was conducted using a HOPE-MED 8050 inhalation exposure system (HOPE Biotechnology, Tianjin, China) with 20 commercial cigarettes (Daqianmen brand, containing 0.8 mg of nicotine, 12 mg of CO, and 10 mg of tar per cigarette) each session [13]. Additionally, on days 1, 11, and 21, these mice received a 0.3 ml intraperitoneal injection of 10% CSE. The control and bexarotene groups were exposed to fresh air and received a 0.3 ml intraperitoneal injection of phosphate-buffered saline (PBS) (VivaCell, Shanghai, China) on days 1, 11, and 21. Mice in the bexarotene and model + bexarotene groups were administered an intraperitoneal injection of bexarotene (Selleck Chemicals, 1 mg/kg, dissolved in a 1:9 ratio of dimethyl sulfoxide to normal saline) 30 min before each daily CS exposure [14]. Mice in the other groups received an equivalent volume of solvent via intraperitoneal injection. All animal experiments were conducted with the approval of the Ethics Committee for Animal Use and Care at Shengjing Hospital under protocol number 2022PS202K.
Fig. 1
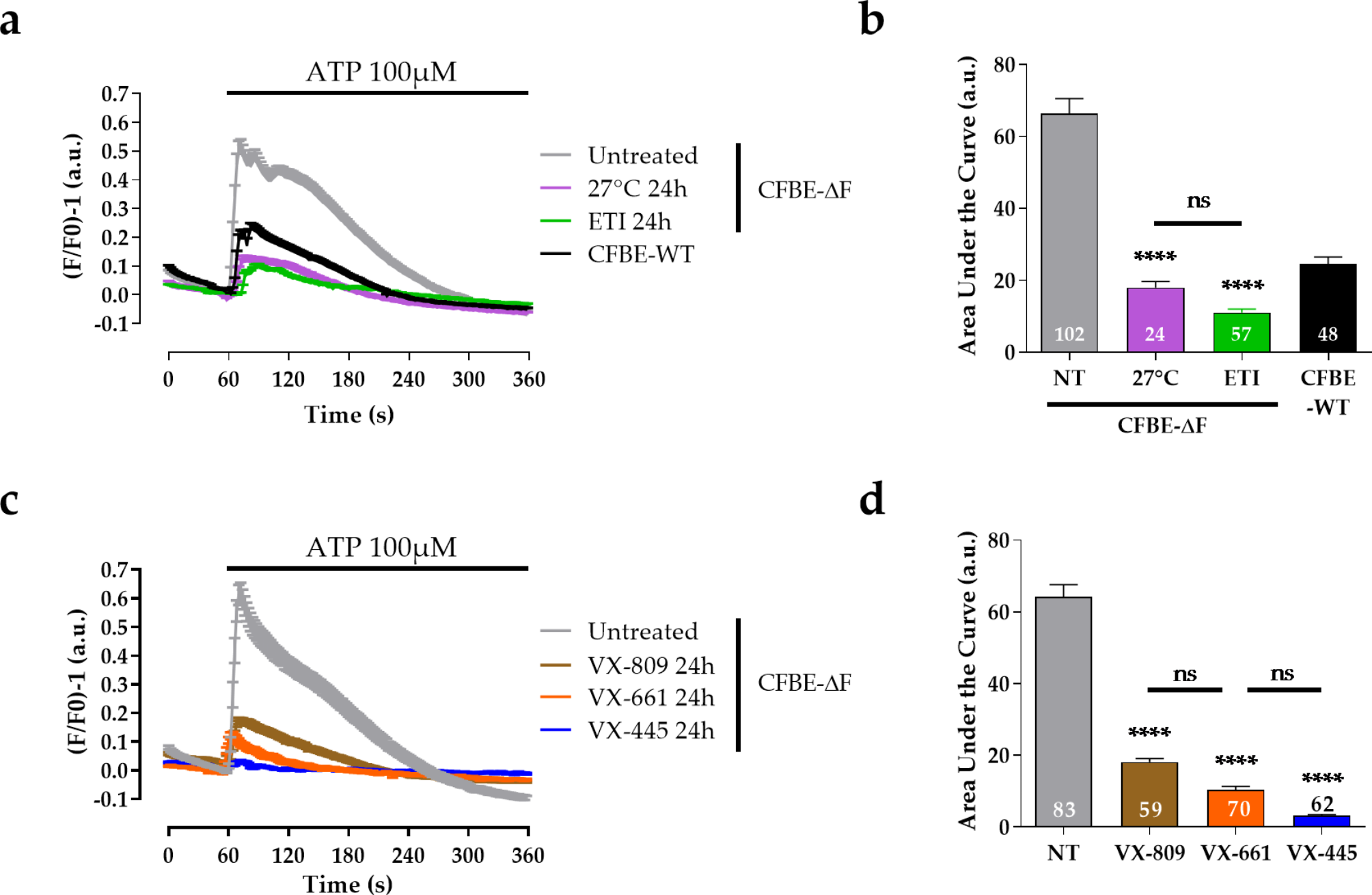
Experimental grouping and emphysema assessment. C57BL/6 mice were divided into four groups: control, model, bexarotene, and model + bexarotene (A). Lung tissue histopathology was analyzed via H&E staining at 100x magnification. The mean linear intercept (MLI) and destructive index (DI) were quantified across the groups (B). The data are presented as the means ± SDs of six replicates. The experiments were conducted three times. Statistical significance is denoted by * for p < 0.05 compared to the control group and # for p < 0.05 compared to the model group
Lung tissue morphometric analysis and inflammatory scoreThe lungs of the mice were perfused with 4% paraformaldehyde at 25 cm H2O pressure and subsequently fixed in the same solution for 48 h. The tissues were then embedded in paraffin, and 4 μm thick sections were prepared for staining. These sections were stained with hematoxylin-eosin (HE) and imaged using light microscopy. The mean linear intercept (MLI) was measured across 10 random fields per sample to assess emphysema severity [7]. The destructive index (DI) was calculated based on the method described [15], involving counting the number of destroyed versus normal alveoli under microscopy, and calculating the DI as follows: DI = (number of destroyed alveoli/total number of alveoli) × 100%. Pulmonary inflammation was evaluated using a 4-point scale (1: minimal, 2: mild, 3: moderate, 4: severe) across four criteria: alveolar septal, peribronchiolar, perivascular infiltrates, and bronchus-associated lymphoid tissue hyperplasia. The scores from each criterion in every sample were aggregated to quantify inflammation [8].
Total and differential cell counts in BALFBronchoalveolar lavage was performed with 1 mL PBS. The bronchoalveolar lavage fluid (BALF) was centrifuged, and cell pellets were resuspended in PBS. Total cell count was determined using a hemocytometer. For differential counts, 100 µL of the cell suspension was placed on glass slides, centrifuged at 1000 rpm for 10 min, and subsequently stained with Wright‒Giemsa for microscopic examination.
ELISA for measurement of TNF-α, IL-1β, and IL-6For the measurement of the cytokines TNF-α, IL-1β, and IL-6, the supernatant from the BALF was collected after centrifugation and stored at -80 °C. The levels of these cytokines were quantified using an enzyme-linked immunosorbent assay (ELISA) following the manufacturer’s protocol (Boster, Wuhan, China). Optical density (OD) values at 450 nm were measured using an Infinite® 200 PRO spectrophotometer (Tecan Trading AG, Switzerland).
Immunohistochemistry evaluation of iNOS and CD206To assess the polarization status of lung tissue macrophages, the M1 and M2 polarization markers iNOS and CD206, respectively, were detected via immunohistochemistry as described in our previous study [7, 8]. Lung tissue sections were incubated with primary antibodies against iNOS (ab15323) at a 1:200 dilution and CD206 (ab300621) at a 1:1000 dilution (both from Abcam, UK) overnight at 4 °C. Subsequently, the sections were treated with horseradish peroxidase-conjugated secondary antibodies (Maixin, Fuzhou, China) at a 1:5000 dilution for 2 h at room temperature. DAB was used for visualization of positive cells, followed by counterstaining with hematoxylin. Images were captured using a light microscope at 400x magnification, and positive cells were quantified using Image-Pro Plus 6.0. Four random fields per section and five sections per animal were analyzed independently by two well-trained investigators. To ensure inter-rater reliability, they used the same equipment and performed the double-blind assessments under the same conditions. If there was a significant discrepancy (greater than 15%) in the evaluation of a sample between the two raters, the sample would either be discarded or reassessed.
Flow cytometryThe BALF was centrifuged at 300 × g for 10 min, and the resulting cell pellet was prepared for flow cytometry. Each tube contained 1 × 106 cells, and Fc receptors were blocked using FcR Blocking Reagent (Miltenyi Biotec, Germany). The cells were then incubated with a FITC-labeled F4/80 monoclonal antibody (11-4801, eBioscience, USA) at 4°C for 40 min in the dark. For intracellular staining of iNOS and CD206, the Fix and Perm Kit (BD Bioscience, USA) was utilized per the manufacturer’s instructions, followed by incubation with PE-labeled iNOS (12-5920) and APC-labeled CD206 (17-2061) monoclonal antibodies (eBioscience, USA) at 4°C for 40 min in darkness. All antibodies were used at the concentrations recommended by the manufacturer. Cells were acquired using a BD Celesta instrument, with F4/80-positive cells gated as AMs and at least 1 × 104 events gated for each sample, consistent with our previous methodology. Data analysis was conducted using FlowJo software (Tree Star, Ashland).
Preparation of the CSECSE was prepared as follows [16]: Smoke from two unfiltered Daqianmen cigarettes (0.8 mg nicotine, 12 mg CO, 10 mg tar each) was drawn into 20 ml RPMI 1640 medium using a vacuum pump. The extract was filtered through a 0.22-µm filter to remove particles and bacteria, then standardized to a 10% concentration. OD measurements at 320 nm ensured consistency in CSE preparation.
Cell culture and drug treatment in vitroMH-S cells (Pricella Life Science, Wuhan) were cultured in RPMI 1640 with 10% FBS, penicillin (100 U/mL), and streptomycin (100 µg/mL). Cells were passaged at 70-80% confluence using a 1:3 ratio. For the experiments, MH-S cells were exposed to 1% CSE for 8 h to assess their polarization status and PPARγ/HO-1 expression. Cells were treated with bexarotene at concentrations of 500 nM and 1 µM one hour prior to CSE exposure. To examine the roles of RXRα, PPARγ and HO-1, cells were pretreated with a RXRα inhibitor (HX531, 1 µM; Cayman, USA), a PPARγ inhibitor (GW9662, 10 µM; Cayman, USA), a PPARγ inhibitor (rosiglitazone, 30 µM; Cayman, USA) and an HO-1 inhibitor (tin protoporphyrin IX dichloride [SnPPIX], 50 µM; MedChemExpress, USA) one hour before CSE or bexarotene application.
Cell viability assayCell viability was evaluated using the CCK-8 assay, as per the manufacturer’s guidelines. The cells were plated in quintuplicate in 96-well plates (Nest Biotechnology, Wuxi, China). Cells were treated with 0, 50 nM, 100 nM, 200 nM, 500 nM, 1 µM, 2 µM, 5 µM or 10 µM bexarotene for 12 h, or treated with 1 µM bexarotene for 2 h, 4 h, 8 h, 12 h and 24 h. Subsequently, 10 µL of CCK-8 reagent was added to each well. The plates were incubated at 37 °C for 2 h, after which the absorbance at 450 nm was measured. Cell viability is expressed as a percentage of that of the control group.
PPARγ transcription activity detectionThe nuclear protein of MH-S cells was extracted with a Nuclear Extraction Kit (Cayman, No. 10009277, USA). The transcriptional activity of PPARγ was detected with a PPARγ Transcription Factor Assay Kit (Cayman, No. 10006855, USA) according to the manufacturer’s protocol. The absorbance at 450 nm was determined and used to calculate the transcriptional activity of PPARγ.
Real-time qPCR assayTotal RNA was extracted using the Total RNA Extraction Kit (Promega, USA) following the manufacturer’s instructions, achieving an OD 260/280 ratio between 1.8 and 2.0. cDNA synthesis was performed with a reverse transcription kit (Takara, China). Primers for iNOS, TNF-α, Arg-1, CD206, and β-actin were designed using Primer Premier 5.0 software, with the following sequence:
iNOS forward: TGGAGCGAGTTGTGGATTGT.
iNOS reverse: GTAGTGATGTCCAGGAAGTAGGT.
TNF-α forward: CCACGCTCTTCTGTCTACTGA.
TNF-α reverse: GATGATCTGAGTGTGAGGGTCT.
Arg-1 forward: GAGAAGGTCTCTACATCACAGAAG.
Arg-1 reverse: TTCACAGTACGAGTCACCTCC.
CD206 forward: GTTTCCATCGAGACTGCTGC.
CD206 reverse: GCCACTTTCCTTCAACATTTCG.
β-actin forward: ACGGTCAGGTCATCACTATCG.
β-actin reverse: GTTTCATGGATGCCACAGGATT.
Quantitative PCR was conducted using a qPCR kit (Takara, China) on a Roche 480 LightCycler (Basel, Switzerland). The protocol included an initial denaturation at 95 °C for 30 s, followed by 40 cycles of 95 °C for 5 s and 60 °C for 30 s. Gene expression levels were quantified using the 2-ΔΔ CT method and normalized to β-actin expression.
Western blottingMH-S cells were lysed using RIPA buffer supplemented with a phosphatase inhibitor cocktail and 1 mM PMSF. The lysates were sonicated and then centrifuged at 12,000 × g for 30 min. The protein concentrations in the supernatants were determined using the BCA method. Proteins (30 µg) were resolved on a 10% polyacrylamide gel and subsequently transferred to a polyvinylidene fluoride membrane. The membrane was blocked with milk and then incubated overnight at 4 °C with primary antibodies at a dilution of 1:1000. Primary antibodies against PPARγ (16643-1-AP), HO-1 (10701-1-AP), and GAPDH (10494-1-AP) were obtained from Proteintech (Wuhan, China). The following day, the membrane was incubated with HRP-conjugated secondary antibodies at room temperature for 2 h. Protein bands were visualized using enhanced chemiluminescence (New Cell & Molecular Biotech, China) and analyzed with ImageJ software.
Statistical analysisThe results are presented as the mean ± standard deviation. Data analysis was conducted using SPSS (version 23.0). Differences between groups were assessed using either Student’s t test or one-way ANOVA. The experiments were replicated three times, and a p value less than 0.05 was considered to indicate statistical significance.
留言 (0)