
記住我






It is known that there are important distinctions between pediatric and adult patients with APL [19]. Based on the circumstantial evidence, we hypothesize that induction therapy with additional one dose of anthracycline to ATRA-arsenic combination can reduce the incidence of leukocytosis and DS, the time for achieving HCR, and hospital days required for inpatient management in pediatric patients with NHR APL.
Eligibility criteriaPatients will be recruited from 29 hospitals in South China enrolled in SCCCG-APL study. Information of the hospitals enrolled are listed in Supplementary Table 1. All study participants will receive detail information about the study and the treatments and sign informed consent before participation. The participants will be able to withdraw from the study at any time without giving reasons.
Inclusion criteriaParticipants fulfilling the following:
1. Age ≤ 16 years.
2. Newly diagnosed APL with PML-RARα/t (15; 17).
3. Initial peripheral blood WBC ≤ 10 × 109/L (NHR APL).
Exclusion criteriaParticipants meeting one or more of the following:
1.Death from any cause before genetic diagnosis and randomization
2.Coma, convulsion, paralysis due to intracranial hemorrhage, cerebral thrombosis, or central nervous system leukemia before genetic diagnosis and randomization
3.Prolonged QT syndrome at diagnosis
4.Declined randomization
Sample size calculationBased on the previous studies [3] assuming 80% hyperleukocytosis rate in the control (ATRA-RIF) group and conservatively assuming 45% hyperleukocytosis rate in the experimental (ATRA-RIF plus chemotherapy) group, 2.5% type I error (one-sided), and 80% power, we used PASS 11 to calculate a required sample size of 30 evaluable patients per group to detect a between-group difference. Allowing for a withdrawal rate of 5% would require 32 patients per group. Therefore, we plan to enroll 64 patients into two groups (1:1).
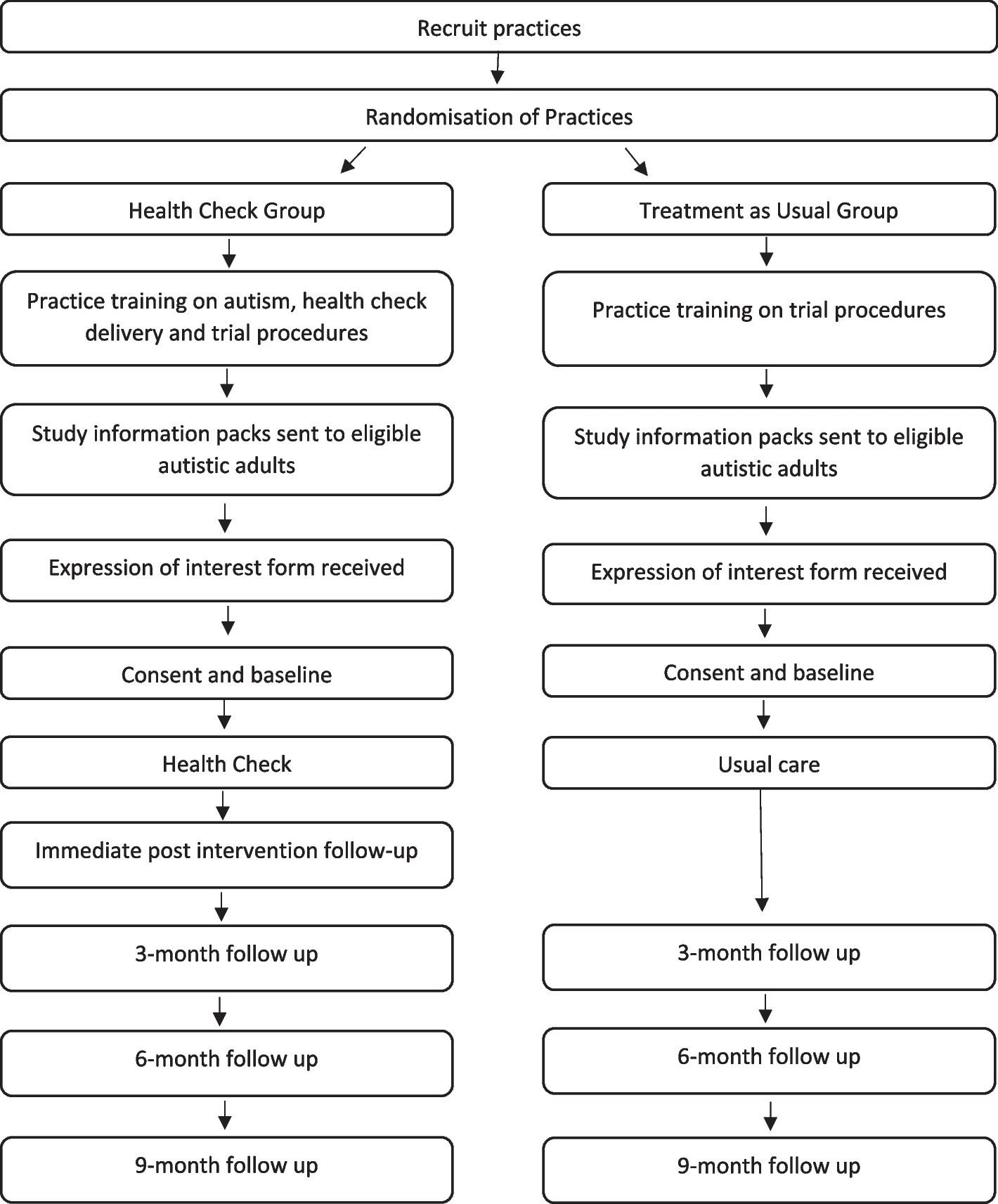
RandomizationOnce informed consent is obtained, participants will be randomly assigned (1:1) by a computer-generated, random-allocation schedule to one of two groups: the ATRA-RIF plus chemotherapy group (experimental group) and ATRA-RIF group (control group) (Fig. 1). A clinical research coordinator, operating independently, will allocate patients to either group. The creation of the allocation sequence for participants will be overseen by a second researcher.
Fig. 1 Blinding
BlindingBlinding method will not be used in this study because it is not feasible for this study for patients and the research team. Both patients and researchers are informed of the grouping.
InterventionsChildren with NHL APL will be randomly assigned by a computer-generated, random-allocation schedule to one of two groups: ATRA-RIF plus chemotherapy group (experimental group) and ATRA-RIF group (control group) (Figs. 2 and 3).
Fig. 2 Fig. 3
Fig. 3
Treatment groups. HCR, hematological complete remission; MRD, minimal residual disease; IT, intrathecal injections; 6MP, 6-Mercaptopurine
Induction therapyInduction therapy contains ATRA and RIF, and addition of one dose of idarubicin (IDA) for patients in experimental group. ATRA will be administrated once the diagnosis of APL is suspected based on morphological features. RIF will be given when the diagnosis of APL is genetically confirmed (5–6 days later from the day of morphological diagnosis).
The control group will receive oral ATRA at 25 mg/m2/day from day 1 until day 42 or HCR and oral RIF at 0.135 g/kg/day from day 5 until day 42 or HCR.
The experimental group will receive the same treatment as those assigned to the control group, with addition of IDA at 10 mg/m2 administrated on day 2.
Adverse events during induction treatment should be monitored mainly including coagulopathy, leukocytosis, DS, and QT prolongation. Blood routine and/or coagulation test should be performed every day or twice a day if there are leukocytosis and/or coagulopathy. Liver and kidney function tests and electrocardiogram should be performed every week.
At the initiation of and during induction therapy, when patients’ WBC count was over 10 × 109/L, hydroxyurea (100 mg/kg/day) was administered until WBC < 10 × 109/L. Dexamethasone (0.3 mg/kg/day) was given if differentiation syndrome or ATRA-associated pseudotumor cerebri was suspected. The use of heparin or low-molecular weight heparins for management of coagulopathy was not mandatory but encouraged because ATRA might increase thrombosis risk [20,21,22]. Heparin was given at a low dose of 0.5–1 mg/kg intravenously for 24 h daily if indicated until the coagulopathy resolved.
Transfusions of platelet and fresh-frozen plasma, cryoprecipitate, and/or human fibrinogen were given for the aims of maintaining platelet counts greater than 30 × 109/L and fibrinogen greater than 1.5 g/L, respectively.
Consolidation therapyConsolidation will start when patients achieve HCR. Patients receive three courses of consolidation therapy every 4 weeks. Each consolidation consists of ATRA (25 mg/m2/day, d1-d21) and RIF (0.135 g/kg/day, d1-d21). Intrathecal injections (IT) are administered on day 1 of each course (Table 1). Blood routine, liver and kidney function tests, and electrocardiogram should be performed before the beginning of each consolidation.
Table 1 Drug dose for intrathecal injectionMaintenance therapyCycle 1: ATRA (25 mg/m2/day, weeks 1–3), RIF (0.135 g/kg/day, weeks 1–3), MTX (20 mg/m2/week, weeks 1–12), 6-mercaptopurine (6MP, 50 mg/m2/day, weeks 1–12).
Cycle 2: ATRA (25 mg/m2/day, weeks 1–3), MTX (20 mg/m2/week, weeks 1–12), 6MP (50 mg/m2/day, weeks 1–12).
Cycles 1 and 2 are repeated for a total of 6 cycles.
Relevant concomitant care permitted or prohibited during the trialParticipants will continue to receive standard care and treatment from a specialized physician throughout the study. There are no limitations on appropriate concurrent care for patients, who are also permitted to sustain their routine healthcare regimen.
Baseline characteristicsBaseline characteristics in each group will be analyzed using descriptive statistics, including means or medians for continuous variables and percentages for categorical variables.
EventsEvery case of hyperleukocytosis, DS, coagulopathy events, infection, gastrointestinal reaction, pseudotumor cerebri, and treatment-related toxicity of the liver, kidney, and heart occurring during the study must be recorded. The following information will be recorded: occurrence time, severity, duration, adopted measure, and the outcome of the adverse event.
Any serious adverse events occurring during the study period should be documented. If the serious events occur, investigators should determine whether participants should be withdrawn from the study depending on the patient’s condition and must take the necessary steps immediately and report to the ethics committee. Serious adverse events that are still ongoing at the end of the study must be followed up to determine the final outcome. Additionally, participants have the autonomy to discontinue their involvement in the trial at any point and for any reason. Comprehensive explanations for the withdrawal of each participant will be provided in the study results.
Follow-up monitoringMinimal residual disease (MRD) by qRT-PCR will be monitored at following treatment points: at the end of induction and consolidation using bone marrow samples and then every 24 weeks thereafter using blood or bone marrow samples for 120 weeks (48 weeks after maintenance is finished) [23]. If MRD is positive (PML-RARa ≥ 0.01% by qRT-PCR) during or after maintenance therapy, patients will be considered as having molecular relapse of APL, and qRT-PCR will be repeated after 1–2 weeks for confirming the relapse. Hematologic relapse is defined by bone marrow blasts of 5%, reappearance of blasts in the blood, or development of extramedullary disease.
Study end pointsThe primary endpoint is the incidence of hyperleukocytosis, assessed at the end of the induction treatment. The secondary endpoints include the incidence of DS, hospital days of induction, time from diagnosis to HCR, the rate of molecular complete remission (MCR) at end of induction treatment, and the consumption of blood components including the amount of platelets, plasma, and cryoprecipitate used during induction.
Data collection and managementWe will inform patients about the importance of follow-up for health monitoring and early detection of relapse and will maintain regular telephone contact with patients to promote participant retention and complete follow-up. Patient information will be entered into the paper case report forms (CRFs) promptly and synchronously with input into the electronic CRF. Researchers must ensure that the data is true, complete, and accurate. After completing the observation of study cases, the data will be entered into the electronic data record as soon as possible, with entry performed by authorized personnel into the database. We will use the latest version of the Medical Dictionary for Regulatory Activities (MedDRA) to code adverse events (AEs) and medical history and the latest version of the World Health Organization Drug Dictionary (WHODRUG) to code concomitant medications. Additionally, researchers must ensure the privacy rights of patients participating in the clinical trial (anonymity). In all submitted documents, patients’ identities can only be identified using clinical trial identification codes (such as hospitalization numbers). Researchers must also properly safeguard the original documents containing personal information, such as the names and addresses of clinical trial participants. Any unexpected problems during this process should be documented and promptly reported. Furthermore, we will collect demographic and disease characteristics, including sex, age, height, weight, and medical history, to determine whether the two randomized groups are similar.
The ethics committee will oversee the researchers and all facets of the study to ensure strict adherence to ethical principles and the safeguarding of patient health and dignity. Should any ethical breaches occur, corrective measures will be implemented, and the study may be halted if necessary. The committee will monitor the integrity and accuracy of data collection to control its quality. The committee will also decide whether member hospitals would be included in the final analysis based on the authenticity of the data they submit. Any modifications to the study protocol and relevant documents must be submitted to the ethics committee for review and can only be implemented after obtaining the committee’s approval. The serious adverse event (SAE) and suspected unexpected serious adverse reaction (SUSAR) reports, as well as protocol violation reports, must be submitted to the ethics committee in a timely manner. Specifically, SAEs at this center must be reported within 24 h, while SAEs at other centers must be reported at least every 3 months; SUSARs must be reported at least every 6 months. Additionally, protocol violations must be reported to the ethics committee within 1 month of discovery. Annual reports should be submitted every year, and if the clinical study is suspended or terminated early, a discontinuation report must be submitted.
Data sharing is not applicable to this protocol paper as no data sets have been generated or analyzed in current. The results should be made public within 24 months of the end of the study. The end of the study is the time point at which the last data item is to be reported, or after the outcomes of data are sufficiently mature for analysis. A full report of the outcomes should be made public no later than 3 years after the end of the study. Confidential information pertaining to participants will remain undisclosed in all publications. Results will also be available through Chinese Clinical Trials Registry.
Statistical analysisThe analysis populations for this study are defined as follows: the Full Analysis Set (FAS) includes all participants who signed informed consent and will be used for baseline analysis. The Safety Analysis Set comprises all participants who received the SCCCG-APL 2020 protocol and had at least one safety assessment after induction, while the Per-Protocol Analysis Set (PPAS) includes participants who fully adhered to the treatment regimen of either the experimental or control group without any validated protocol deviations and who completed the induction treatment along with minimal residual disease (MRD) testing. Additionally, for the population with protocol deviations, we will conduct an intention-to-treat (ITT) analysis, and multiple imputation will be applied to handle missing data.
Interim analysis is not applicable to this study. Firstly, the planned sample size is adequate to ensure robust statistical power at the conclusion of the study. Interim analyses might not capture the full spectrum of treatment effects, which could lead to premature conclusions and potentially inflate type I error rates. Furthermore, conducting interim analyses could introduce biases, as they may influence the treatment allocation or participants’ behaviors. We wanted to maintain the integrity of the study results without external influences. Additionally, previous study has already demonstrated the safety of anthracycline. Given the nature of our study and the regulatory framework, we believe that a final analysis at the end of the study will provide a clearer and more comprehensive understanding of the treatment’s efficacy and safety.
Statistical analysis will be performed using the SPSS software version 20.0. Descriptive analysis (calculations of averages, frequencies, proportions, or rates) will be conducted. Comparisons will be made using Student’s t test or the Mann–Whitney U test for comparison of continuous variables and Pearson’s χ2 test for dichotomous variables. Student’s t-test will be used to compare normally distributed variables, while Mann–Whitney U test will be used to compare variables with non-normal distribution. The chi-square test or Fisher’s exact test will be used to compare categorical variables between the two groups. Survival functions will be estimated using the Kaplan–Meier method and will be compared using the log-rank test. All statistical tests will be two-sided, and P < 0.05 is statistically significant.
Standard Protocol Items: Recommendations for Interventional Trials (SPIRIT)This protocol has been written in accordance with the SPIRIT guidelines [24]. The SPIRIT checklist is available in Supplementary File 1.
留言 (0)