
記住我


Patents with metastatic pancreatic cancer assessed in this study were included in the CheckPAC (NCT02866383) [7] or TRIPLE-R (NCT04258150) [8] phase 2 clinical trials conducted at the Department of Oncology, Copenhagen University Hospital, Herlev, Denmark. Patients in the CheckPAC study received nivolumab and ipilimumab in combination with stereotactic body radiotherapy (SBRT) [7]. Patients in the TRIPLE-R cohort were treated with nivolumab, ipilimumab, SBRT, and tocilizumab [8]. The clinical trials received approval from the relevant Ethics Committee and were conducted in accordance with the Declaration of Helsinki and Good Clinical Practice. The CheckPAC trial was approved by the Danish Ethics Committee (H-16031247) and the Danish Data Protection Agency (j.nr. 2012-58-0004; HGH-2016-112; I-Suite j.nr. 05088) [7]. The TRIPLE-R trial was approved by the Danish Ethics Committee (H-19087729) and the Danish Data Protection Agency (j.nr. P-2020-398) [8]. All patients provided written informed consent. The following clinical parameters at baseline were available for both studies: sex (female/male), age, Eastern Cooperative Oncology Group (ECOG) performance score (0/1), C-reactive protein (mg/L), number of leukocytes per mL, number of lymphocytes per mL, neutrophil-lymphocyte ratio, albumin (g/L), hemoglobin (mM) and prior treatments (≤2/>2). Peripheral blood mononuclear cells (PBMCs) from patients were isolated and cryopreserved as previously described [9]. The samples used for this study were collected at baseline and 8 weeks posttreatment initiation.
MiceAnimal experiments were carried out at the animal facility of the Department of Oncology, Copenhagen University Hospital, Herlev, Denmark, adhering to guidelines set by the Federation of European Laboratory Animal Science Association (FELASA) and conducted under a license granted by the Danish Animal Experimentation Inspectorate (2021-15-0201-01001). Female C57BL/6 mice, aged 10–20 weeks, were either purchased from Taconic or bred in-house from a C57BL/6JBomTac lineage.
Murine pancreatic cell lines and tumor modelsThe Pan02 cell line was obtained from the cell line biobank at the National Center for Cancer Immune Therapy (Denmark) and cultured in RPMI-1640 GlutaMAX (Gibco) supplemented with 10% heat-inactivated fetal bovine serum (FBS, Gibco) and 1% penicillin/streptomycin (P/S, Gibco). The KPC cell line, which was established from primary tumors from KPCY mice, was purchased from Kerafast (2838c3) and cultured in high-glucose DMEM with GlutaMAX pyruvate (Gibco) supplemented with 10% FBS and 1% P/S. Cancer cells were expanded in vitro, and then, the mice were subcutaneously (s.c.) inoculated with 5 × 105 cells in the right flank. Upon tumor palpability, the mice were assigned to treatment groups by stratified randomization on the basis of tumor volume. A digital caliper was used to measure the tumor dimensions three times a week. The tumor volume was calculated as 0.5 × length × width^2.
PeptidesThe following murine TGFβ1-derived peptides were used for this study: mTGFβ (mTGFβ)−18–32 (15mer, LLVLTPGRPAAGLST), mTGFβ − 215–223 (9mer, QGFRFSAHC), mTGFβ − 282–289 (8mer, TNYCFSST) and mTGFβ − 334–342 (9mer, TQYSKVLAL), which constitute the TGFβ vaccine [10]. Peptides were dissolved in 20 mM dimethyl sulfoxide (DMSO), with the exception of mTGFβ (mTGFβ)−18–32, which was reconstituted in 2 mM H2O. For experiments with human PBMCs, the TGFβ1-derived peptide TGFβ-15 (REAVPEPVLLSRAELRLLRL) and the Clostridium tetani-derived “tetanus” peptide (AQYIKANSKFIGITEL) were used following reconstitution with 10 mM dimethyl sulfoxide (DMSO). Peptides were purchased from Schäfer (>90% purity).
TreatmentsThe mice were treated with the TGFβ vaccine as previously described [10]. In brief, the mice were vaccinated s.c. at the base of the tail, with two emulsions per treatment point. One contained 100 µg of the murine TGFβ1-derived peptide mTGFβ-18–32. The other group comprised 50 µg of each of the following peptides: mTGFβ − 215–223, mTGFβ − 282–289, and mTGFβ − 334–342. The vaccines were formulated by creating an emulsion of the peptide solution with Montanide ISA 51 VG (Seppic) at a 1:1 ratio. For control vaccinations, mice were treated s.c. at the base of the tail with an emulsion generated by emulsifying water with Montanide ISA 51 VG at a 1:1 ratio. For the tumor studies, the mice were vaccinated when the tumors emerged (day 8 postinoculation) and 7 days later (day 15 postinoculation). For cocultures with T cells from tumor-free, vaccinated mice, vaccinations were performed on days 0 and 7. To block IL-6R in vivo, the InVivoMAb anti-mouse IL-6R blocking antibody was purchased from BioXCell (clone 15A7). Each mouse received 200 µg of antibody in 50 µL of PBS s.c. next to the tumor per mouse on day 8 postinoculation and every 3rd, 4th day for a total of six injections. As a control, 50 µL of PBS was administered at the same location and following the same schedule.
Generation of tumor-conditioned mediaTumor-conditioned media (TCM) was generated as previously described [10]. Briefly, Pan02 tumor-bearing mice that were either untreated or treated with the TGFβ vaccine were euthanized on days 25–30 postinoculation. Tumors were collected, and single-cell suspensions were generated. A total of 1 × 105 cells were seeded per well in a U-shaped bottom plate. The cell culture supernatant, termed TCM, was collected after a 48 h incubation.
Cell sortingCancer-associated fibroblasts (CAFs) were isolated from KPC tumors. In brief, KPC tumor-bearing mice were euthanized 14–18 days postinoculation. Tumors were harvested, cut into smaller pieces, and digested for 30 min at 37 °C and 300 rpm in digestion buffer (RPMI-1640 GlutaMAX supplemented with 1% P/S, 1 mg/mL collagenase type IV (Sigma‒Aldrich), 75 µg/mL DNase I (Sigma‒Aldrich), and 5 mM CaCl2). CAFs were sorted on the basis of the marker CD90.2 via the Mouse Tumor-Associated Fibroblast Isolation Kit from Miltenyi Biotec, according to the manufacturer´s instructions. T cells were isolated from the spleens of vaccinated mice. Briefly, 1 week after the last vaccination, the mice were sacrificed, and the spleens were harvested. Single-cell suspensions were generated by processing the spleens through a 70 µm cell strainer and lysing red blood cells with RBC lysis buffer (QIAGEN). T cells were sorted with the Mouse Pan T-Cell Isolation Kit II from Miltenyi Biotec following the manufacturer’s instructions. Monocytes were sorted from the bone marrow of untreated, tumor-free mice. In brief, the mice were euthanized, and the femurs and tibias were collected. The bone marrow was harvested by flushing the bones with PBS. Single-cell suspensions were generated by processing the sample through a 70 µm cell strainer. Monocytes were isolated from Miltenyi Biotec via the Mouse Monocyte Isolation Kit (BM) according to the manufacturer´s guidelines.
CoculturesSorted CAFs were cultured in DMEM supplemented with 10% FBS and 2% P/S for in vitro expansion. A total of 8.5 × 104 CAFs were cultured with 1 × 106 T cells (1:12 CAF:T-cell ratio) per well in a final volume of 200 µL of DMEM, 10% FBS, and 1% P/S in a U-shaped bottom 96-well plate. Following a 48 h incubation, the cells were collected for flow cytometry, and the supernatant was saved for enzyme-linked immunosorbent assay (ELISA).
ELISAThe IL-6 concentration in the medium and in the coculture supernatant was determined via mouse IL-6 DuoSet ELISA (R&D Systems, DY406) following the manufacturer´s instructions. The optical density was determined with an Epoch Microplate Spectrophotometer (BioTek Instruments) via Gen5 software.
ELISpotThe presence of peptide-specific T cells in murine spleens or in human PBMCs was assessed via IFNγ enzyme-linked immunospot (ELISpot), as previously described [9, 10]. ELISpot plates (Mabtech) were coated with an unconjugated anti-IFNγ antibody (Mabtech, AN18 for mouse, 1-D1K for human) diluted to 12 µg/mL or 7.5 µg/mL for mouse or human, respectively, in PBS and incubated overnight. A total of 8 × 105 murine splenocytes in 200 µL of RPMI-1640 supplemented with 10% FBS, 1% P/S, or 2 × 105 PBMCs in 200 µL of X-VIVO 15 (Life Science) were plated per well in triplicate. Human PBMCs were stimulated in vitro with TGFβ-15 and cultured for approximately 10 days before being used in the assay, as previously described [9, 22]. Following a 24 h incubation at 37 °C, the plates were rinsed with PBS. The cells were stimulated with peptide solution at a working concentration of 5 µM. The general response to the TGFβ vaccine was evaluated by stimulation with a peptide pool comprising all five peptides that conform the TGFβ vaccine to a working concentration of 5 µM per peptide. Biotinylated anti-IFNγ antibodies (Mabtech, R4-6A2 for mouse, 7-b6-1 for human) diluted to 1 µg/mL or 0.75 µg/mL for mouse or human, respectively, in ELISpot buffer (PBS, 0.5% bovine serum albumin, and NaN3) were used as secondary antibodies. The plates were incubated for 2 h at room temperature, followed by a PBS-washing step. Streptavidin-ALP (Mabtech) diluted 1:1000 in ELISpot buffer was added, and the plates were incubated for 1 h at room temperature. Unbound enzyme was washed off with PBS. The assay was developed following a 1–5 min incubation at room temperature with the enzyme-substrate BCIP/NBT (Mabtech). The reaction was stopped with tap water. A CTL ImmunoSpot S6 Ultimate-V analyzer with ImmunoSpot software (v5.1) was used to count the spots. Specific responses are expressed as the difference between the average number of spots in peptide-stimulated wells and those in unstimulated wells. TGFβ-15-specific responses at baseline were classified as high (TGFβ-15high) or low (TGFβ-15low) following the cutoff of 50 specific cells per 250,000 plated cells, as described by Mortensen et al. [9]. We assessed TGFβ-15-specific and tetanus-specific responses in all 25 patients (10 with TGFβ-15high and 15 with TGFβ-15low) included in the TRIPLE-R study [8] from whom PBMCs at baseline were available.
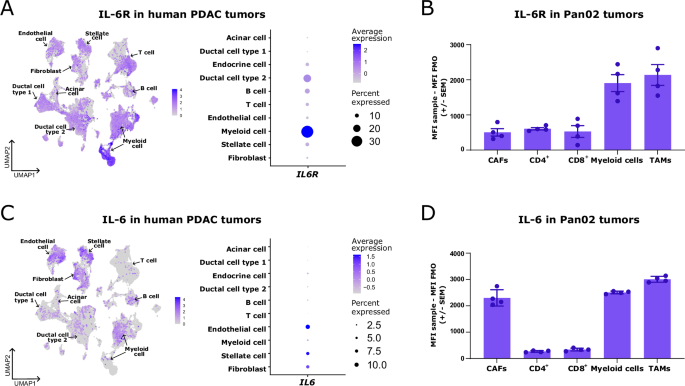
Flow cytometryZombie Aqua (BioLegend, 423101) was used as a viability marker for flow cytometry of murine samples. Fc receptors were preblocked with mouse FcR blocking reagent (1:10; Miltenyi Biotec). The following antibodies were used to assess the mean fluorescence intensity (MFI) of IL-6 and IL-6R in CAFs from untreated Pan02 tumors: CD45-FITC (BioLegend, 103108), CD90.2-BV605 (BioLegend, 140317), CD8-BB700 (BD Pharmingen, 566409), CD4-BV421 (BioLegend, 100438), CD11b-APC/Cy7 (BioLegend, 101226), F4/80-PE (BioLegend, 123110), CD126 (IL6Ra)-APC (BioLegend, 160408) and IL-6-APC (BioLegend, 504508). CAFs were gated as live CD45− CD90.2+ cells. CD4+ T cells were gated as live CD45+ CD4+ cells. CD8+ T cells were gated as live CD45+ CD8+ cells. Myeloid cells were gated as live CD45+ CD11b+ cells. Tumor-associated macrophages (TAMs) were gated as live CD45+ CD11b+ F4/80+ cells. The following antibodies were used to evaluate changes in the IL-6 MFI in CAFs after culture with T cells: CD45-PE/Cy7 (BioLegend, 103114), CD90.2-BV605 (BioLegend, 140317) and IL-APC (BioLegend, 504508). CAFs were gated as live CD45− CD90.2+ cells. The following antibodies were used to investigate changes in the T-cell compartment in Pan02 tumors across treatment groups: CD45-BV605 (BioLegend, 103140), CD3-AF700 (BioLegend, 100216), CD8-BB700 (BD Pharmingen, 566409), CD4-BV421 (BioLegend, 100437), FoxP3-APC (eBioscience, 17-5773-82), CD25-BV786 (BD Horizon, 564023) and PD-1-PE (BioLegend, 109104). T cells were gated as CD45+ CD3+ T cells. CD4+ T cells were gated as CD45+ CD3+ CD4+ T cells. CD8+ T cells were gated as CD45+ CD3+ CD8+ T cells. Regulatory T cells (Tregs) were gated as CD45+ CD3+ CD4+ Foxp3+ CD25+ cells. The following antibodies were used to assess changes in the myeloid subset in Pan02 tumors across treatment groups and changes in the phenotype of BMDMs differentiated in the absence or presence of IL-6: CD45-BV605 (BioLegend, 103140), CD3-AF700 (BioLegend, 100216), CD11b-Pacific blue (BioLegend, 101223), F4/80-FITC (BioLegend, 123108), MR (CD206)-PE/Cy7 (BioLegend, 141719), MHC-II-APC/Cy7 (BioLegend, 107627), Arg1-PE (R&D Systems, IC5868P), PD-L1-APC (BD Biosciences, 564715) and CD8a-PerCP/Cy5.5 (Pharmingen, 551162). Myeloid cells were gated as live CD45+ CD3− CD11b+ cells. TAMs or BMDMs were gated as live CD45+ CD3− CD11b+ F4/80+ cells.
Characterization of human PBMCs with flow cytometry at baseline (w0) and 8 weeks posttreatment (w8) was performed on all patients from whom PBMCs were available at both timepoints (n = 10 patients for the CheckPAC trial and n = 9 for the TRIPLE-R trial). NIR (Invitrogen, L34993) was used as a live marker for flow cytometry with human PBMCs. The following antibodies were used to assess changes in the T-cell compartment in human PBMCs: CD3-BV786 (BD Biosciences, 563800), CD8-PE-CF594 (BD Biosciences, 566850) and CD4-BV510 (BD Biosciences, 562970). T cells were gated as live CD3+ cells. The following antibodies were used to assess changes in myeloid subsets in human PBMCs: CD3-BV786 (BD Biosciences, 563800), CD14-PE-CF594 (BD Biosciences, 562335), CD16-BV650 (BD Biosciences, 563692), HLA-DR-PerCP-Cy5.5 (BD Biosciences, 560652), CD123-BV605 (BD Biosciences, 564197), CD1c-APC (BioLegend, 331524), CD11c-PE (BioLegend, 371504), CD33-BV510 (BD Biosciences, 563257), CD56-FITC (BD Biosciences, 345811), NKG2a-BV421 (BD Biosciences, 747924), and CD19-BV711 (BD, 563038). Monocytic populations were identified via high-dimensional analysis of flow cytometry data from human PBMCs with Cytobank. For the staining of intracellular proteins, the samples were fixed and permeabilized with eBioscience Fixation/Permeabilization Concentrate, Diluent, and 10X Buffer (Invitrogen) following the manufacturers’ instructions. Data acquisition was performed on an ACEA NovoCyte Quanteon (Agilent), and the data were analyzed via FlowJo V.10.6.1 (Tree Star).
High-dimensional data analysis of flow cytometry dataHigh-dimensional analysis of flow cytometry data from human PBMCs stained with a myeloid panel and murine tumors stained with a myeloid panel was performed via Cytobank (https://cytobank.org) [39]. For data from human PBMCs, uniform manifold approximation and projections (UMAP) analysis was performed on manually gated live CD3− cells, with 50,000 events per sample, 15 neighbors, a minimum distance of 0.01 and collapsed outliers (Z score > 3) with normalized scales. The following markers were used to generate the UMAP: CD14, CD16, HLA-DR, CD123, CD1c, CD11c, CD33, CD56, NKG2a and CD19. The data were then fed into the FlowSOM clustering algorithm, which samples 50,000 events per sample. Hierarchical consensus clustering with 10 iterations and 100 clusters was used after scale normalization, resulting in a total of 30 metaclusters. Metaclusters were annotated on the basis of median fluorescence intensity (MFI) heatmaps generated in Cytobank. Clusters with less than 1% frequency were excluded from the analysis, and clusters that would receive the same annotation on the basis of heatmap marker expression were combined if one of them constituted <5% of the live cells. The CD14+ CD16− HLA-DR- clusters were annotated as HLA-DR− classical monocytes, the CD14+ CD16− HLA-DRmid clusters were annotated as HLA-DRmid classical monocytes, the CD14+ CD16+ HLA-DRhi clusters were annotated as monocytes in an intermediate state, and the CD14− CD16+ clusters were annotated as nonclassical monocytes. Clusters corresponding to the monocyte population (n = 4) were selected for downstream analysis. For data from murine tumors, UMAP analysis was performed on manually gated live CD45+ CD3− cells, sampling 1930 events per sample, with 15 neighbors and a minimum distance of 0.01. The following markers were used to generate the UMAP: CD11b, F4/80, MR (CD206), ARG1, MHC-II and PD-L1. The data were then fed into the FlowSOM clustering algorithm, which samples 1930 events per sample. Hierarchical consensus clustering with 10 iterations and 49 clusters was used after scale normalization, resulting in a total of 6 metaclusters. Metaclusters were annotated on the basis of MFI heatmaps generated via the pheatmap (V1.0.12) R package. CD11b− clusters were annotated as non-myeloid cells. CD11b+ clusters were annotated as myeloid cells. F4/80+CD11b+ clusters were annotated as macrophages. Macrophage clusters characterized as MRlow ARG1low MHC-II+ or MRhigh ARG1high MHC-II- were annotated as proinflammatory or anti-inflammatory macrophages, respectively.
ImmunofluorescencePancreases were fixed in 4% paraformaldehyde (PFA) for 24 h at room temperature (RT) and subsequently incubated with 70% ethanol (VWR Chemicals; 20824.365) until paraffin embedding. Sections (4 μm) were mounted on Superfrost Plus slides (Fisher Scientific; 10149870). The paraffin-embedded samples were rehydrated with xylene and ethanol with increasing concentrations of water. After deparaffinization, 0.5% PBS-Tween-20 was used in all washing steps. Antigen retrieval was performed by boiling for 20 min with Tris-EDTA buffer at pH 9.0. The samples were blocked with 1% normal donkey serum (Jackson ImmunoResearch, 017-000-121) and later incubated with primary antibodies (rabbit anti-Keratin7 (Abcam, ab181598, 1:200) and goat anti-IL6 (Santa Cruz, sc-1265, 1:200)) overnight at 4 °C. The sections were then incubated with secondary antibodies (Cy3 donkey anti-rabbit (Jackson ImmunoResearch, 711-166-152, 1:500), FITC donkey anti-goat (Jackson ImmunoResearch, 705-546-147, 1:500)) and DAPI (Sigma‒Aldrich; D9542) for 1 h at room temperature. The samples were mounted with Vectashield® Mounting medium (Vector Laboratories; H-1000). An Olympus® ScanR screening microscope was used for imaging. Image processing and quantification were performed via QuPath software (v.0.4.0) (https://qupath.github.io/). For each sample, 2 or 3 cuts at different depths were stained and imaged (9 tiles per cut). Tiles with staining artifacts or a low number of cells were excluded from the analysis. The cells were detected with the built-in “Cell detection” tool using DAPI. Positive cells were classified with the “Set intensity classification” built-in tool.
RNA extractionRNA was isolated from bone marrow-derived macrophages and Pan02 tumors. The macrophages were allowed to stand down, the supernatant was removed, and the pellet was stored at −80 °C. Pan02 tumors (≤20 mg) were stored in RNAlater (Invitrogen) at −80 °C. For RNA extraction from Pan02 tumors, the material was transferred to RLT buffer (QIAGEN) and mechanically homogenized via a Tissue Lyser (QIAGEN). RNA extraction was performed via the RNeasy Plus Mini Kit (QIAGEN) according to the manufacturer’s protocol. The RNA concentration was quantified with a NanoDrop 2000 Spectrophotometer (Thermo Scientific).
qRT‒PCRComplementary DNA (cDNA) synthesis from 1 µg of RNA was performed via an iScript cDNA synthesis kit (Bio-Rad) following the manufacturer’s instructions. cDNA was diluted 1:3. Reverse transcription‒quantitative PCR (RT‒qPCR) was performed in technical triplicate in an AriaMX Real-Time PCR System with a LightCycler 480 Probes Master (Roche Diagnostics) and the following TaqMAn gene expression assay probes (Life Technologies): Hprt1 (Mm00446968_m1), MCH-I H2-D1/K1 (Mm04208017_mH), MHC-II H2-Ab1 (Mm00439216_m1), Ccl6 Mm01302419_m1), Ccl8 (Mm01297183_m1), Ccl9 (Mm00441260_m1), Pf4(Mm00451315_g1), Alox12e (Mm00521331_m1), Fcer1a (Mm00438867_m1) and Arg1 (Mm00475988_m1). The data were normalized to Hprt1 as a housekeeping gene and analyzed via the 2−dCT method.
RNA sequencingBulk RNA sequencing (RNAseq) was performed as previously described10. Briefly, 500 ng of RNA from Pan02 tumors with an RNA integrity number (RIN) score above 7 was enriched for polyadenylated mRNA via Oligo dT beads (NEBNext). The subsequent steps included fragmentation, random-primed cDNA synthesis (NEBNext), PCR-mediated indexing (NEBNext), size selection, and quantification (KAPA, Roche). The Illumina NovaSeq 6000 platform was utilized for cDNA library paired-end sequencing. The alignment to the GRCm39 mouse reference genome and quantification of reads were conducted following previously described procedures10, employing STAR (V.2.7.9), featureCounts within the subread package (V.2.0.3), and Ensembl gene transcripts (GRCm39.104.gtf). The RNAseq counts were VST (variance stabilizing transformation)-normalized. Differential gene expression was performed via the DESeq2 package (V.1.30) with a cutoff of adjusted p-value < 0.05 and an absolute log2-fold change >0.585. The RNA-seq data have been deposited in the GEO repository under the accession number GSE263363. The enhanced Volcano R package (V.1.8.0) was used to generate volcano plots. Gene Ontology (GO) analysis for biological processes of the differentially expressed genes was performed via the enrichGO functions of the cluster profile (V.4.6.2) package in R. GO terms related to cancer immunity was selected and manually classified into the following 10 categories: myeloid cell chemotaxis/migration, lymphoid cell chemotaxis/migration, response to cytokines/chemokines, phagocytosis, B-cell function, and immunoglobulins, immune activation and immune processes, myeloid immunity, regulation of cytokinesis, metabolism, ECM, and categories related to cancer immunity. GO terms were organized into a network and subsequently plotted via the emapplot function of the enrich plot (V.1.18.4) package in R. Cell-type deconvolution of the bulk samples to infer enrichment scores was performed on transcripts per million normalized counts via mMCP-counter [40], implemented in the R package immunedeconv (v2.1.0) [41].
scRNAseq data from public repositoriesSingle-cell RNA sequencing (scRNAseq) analyses were performed via a publicly available scRNAseq atlas of >70 samples and 136,163 cells from human PDAC tumors generated by Chijimatsu et al. [12]. We downloaded the Seurat object containing normalized expression data and the annotated metadata, which was used to show the clusters identifying different cell types. Visualization of the scRNA-seq data was performed via the FeaturePlot and DotPlot functions from the Seurat R package (V.5.0.1).
Generation of bone marrow-derived macrophagesThe mice were sacrificed, and the femurs and tibias were retrieved. The bone marrow was collected by flushing the bones with PBS. Single-cell suspensions were obtained by passing the sample through a 70 µm cell strainer. To generate bone marrow-derived macrophages (BMDMs), 4 × 106 bone marrow-derived cells were cultured in 10 mL of DMEM with 20% FBS and 1% P/S supplemented with 20 ng/mL human M-CSF (PeproTech) in a Petri dish. On days 3 and 5, the medium was completely removed, and 10 mL of fresh medium supplemented with 20 ng/mL human M-CSF was added to each Petri dish. On day 6, the BMDMs were harvested for downstream analysis. To evaluate the effect of IL-6 on BMDMs, IL-6 (PeproTech) was added to the medium at a concentration of 20 ng/mL.
Monocyte migration assayA total of 900 µL of DMEM supplemented with 10% FBS and 1% P/S containing 70 ng/mL CCL2 (PeproTech) was added to each well of a 24-well plate. A 5 µm membrane insert (Millicell Sigma, PTMP24H48) was placed hanging in each well. A total of 1.25 × 105 monocytes isolated from the bone marrow of untreated, tumor-free mice were placed in 200 µL of DMEM supplemented with 10% FBS and 1% P/S inside the insert. The cell cultures were incubated and allowed to migrate for 2 h or overnight at 37 °C. After this time, 800 µL of the media outside the chamber were harvested, passed through, and resuspended in 100 µL of FACS buffer. A total of 90 µL was acquired on an ACEA NovoCyte Quanteon (Agilent), and the number of events was recorded. The results are expressed as the percentage of migrated cells to the initial cell count in the insert.
Cell length measurementBMDM length was determined via QuPath. Microscopy images were imported into the program, and an area of 2100 pixels was randomly selected. Lines across the length of each cell within the selected area were manually drawn. The length of the pixel corresponding to each line was exported.
Data visualizationThe data were visualized via GraphPad Prism (V.8) or the following R packages: ggplot2 (V.3.3.5) and pheatmap (V1.0.12). ggpubr (V.0.6.0), survival (V.3.5.5), ggsurvfit (V.1.0.0), survminer (V.0.4.9) and gtsummary R packages (V.1.7.2).
Statistical analysisFor preclinical data, unpaired, two-tailed t-tests performed in GraphPad Prism (v8) were used to assess statistical significance, unless otherwise stated. Statistical comparisons of the tumor growth curves were conducted with TumGrowth software [42] (https://kroemerlab.shinyapps. io/TumGrowth), default settings, and a Bonferroni adjustment for correction for multiple comparisons. Linear regressions were performed with GraphPad Prism (v8). For human data, paired or unpaired two-tailed t-tests were performed with the ggpubr R package (V.0.6.0). Patient characteristics at baseline across clinical trials were compared via Fisher’s exact test for categorical variables and the Wilcoxon rank sum test or Wilcoxon rank sum exact test for noncategorical variables via the gtsummary R package (V.1.7.2). Survival analyses for all patients in the CheckPAC trial [7] based on TGFβ-15-specific immunity at baseline were previously published [9]. Here, we assessed survival on the basis of TGFβ-15-specific immunity at baseline only for patients in the CheckPAC trial who received radiotherapy, nivolumab, and ipilimumab to enable comparison with those in the TRIPLE-R trial [8]. Survival analysis for the CheckPAC trial was performed on all 17 patients (13 with TGFβ-15high and 4 with TGFβ-15low) for whom survival data and TGFβ-15-specific immunity data at baseline were available. Survival analysis for the TRIPLE-R trial was performed on all 22 patients (10 with TGFβ-15high and 12 with TGFβ-15low) for whom survival data and TGFβ-15-specific immunity data at baseline were available. Overall survival (OS) and progression-free survival (PFS) were assessed via a log-rank test with the survival (V.3.5.5) and ggsurvfit (V.1.0.0) R packages. PFS was determined by investigator assessment according to the RECIST v1.1 guidelines. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. The data are presented as the means ± SEMs unless otherwise stated.
留言 (0)