Human lung tissue sampling
Lung tissue samples for immunohistochemical staining and for isolating primary lung fibroblasts were obtained from the Henan Provincial Chest Hospital. The IPF samples (n = 3) were surgical remnants of biopsies from patients with IPF undergoing pulmonary transplant. Control lung tissue samples (n = 3) were normal histology tissue obtained from normal disease-free margin of lung cancer resection specimens.
Mouse model of BLM-induced pulmonary fibrosis
Fourteen wild-type C57BL/6 N (8-week-old, male) mice were purchased from Vital River Laboratories in Beijing (China). The mice were anesthetized by inhaling 40% isoflurane (diluted in 1, 2-Propanediol, Aladdin, Shanghai, China)), then given 50 µL normal saline (n = 7) or BLM (n = 7) (1.5 U/kg) by intratracheal injected. On day 21, the mice were sacrificed and lung tissue samples were taken.
AAV-mediated in vivo PTPN21 knock down
Mice were intratracheal instilled with 50 µL of pAAV-CMV-PTPN21 shRNA (Sequence of target, CGGAATAGAATGGGATTATAT, virus titer:1.0 × 1012 vg/m L) or pAAV-CMV-NC). One week later, the mice were challenged of BLM (1.5 U/kg) dissolving in saline or equal volume of saline. Each group consisted of 10 mice. The body weight of the mice was measured every other day during the experiment. After challenged of BLM for 14 days, the mice were anesthetized, and bronchoalveolar lavage fluid (BAL) was taken and counted with a cell counter. Subsequently, the lung tissue samples were collected. Total right lung hydroxyproline content was tested by the Hydroxyproline Assay Kit (Sigma-Aldrich, USA), and the experimental procedures followed the instructions. Data were calculated and presented as µg hydroxyproline/g right lung.
A549 cells culture and treatment
Human A549 cells were purchased from the American Type Culture Collection (CCL-185) and propagated in a complete medium of Dulbecco’s modified Eagle’s medium nutrient mixture Ham F-12 (DME/F-12) (Procell, Wuhan, China) with 10% (v/v) FBS, 100 IU penicillin, and 100 µ g/mL streptomycin under standard culture conditions.
For the H2O2 or BLM treatment on A549 cells, the cells were seeded in 6-well plates, when the cell density reached approximately 50% confluence, the cells were treated with H2O2 (50 µM) or BLM (20 mU) for indicated duration.
Quantitative real-time PCR (qRT‑PCR)
Total RNA from A549 cells and mouse lungs was extracted using RNeasy kits (Qiagen) as previously described [19]. cDNA was synthesized using a reverse transcription kit (Promega Corporation, Wisconsin, USA), and the concentration and purity of the cDNA were analyzed using a Nanodrop spectrophotometer. Subsequently, according to the manufacturer’s instructions, qRT-PCR was performed using an SYBR green kit (Yeasen, Shanghai, China) under a Light Cycler 96 fluorescent quantitative PCR system (Roche). ACTB was used as an internal reference, and the data were calculated using the 2−∆∆Ct method. The forward primer (FP) and reverse primer (RP) pairs for PTPN21 used in this study are listed in Table 1. The ACTB primers used are listed in the previous study [19], the Fibronectin 1 (Fn1), Collagen1a1 (Col1a1) and Acta2 primers of mouse used are listed in the previous study [20].
Table 1 Gene specific primer sequence for qRT-PCRPlasmid transfection
For the plasmid transfection, when the A549 cell reached approximately 80% confluence, the overexpression PTPN21 plasmid of pcDNA3.1 + PTPN21 or the empty pcDNA3.1, and knock down plasmid of pcDNA3.1 + U6 + shPTPN21 (Sequence of target, CAACGAATGATGAAAGGTGTA) or empty pcDNA3.1+U6 plasmid were transfected to A549 cells by Lipofectamine3000 (ThermoFisher, USA) according to the manufacturer’s instructions. The amount of plasmid added in 6-well plate, 24-well plate, and 96-well plate was 2.5 µg/ well, 0.5 µg/ well, and 0.1 µg/ well, respectively.
MRC-5 cells culture and treatment
MRC-5 cells were purchased from ATCC and propagated in a complete medium of MEM (Procell, Wuhan, China) with 10% (v/v) FBS, 100 IU penicillin, and 100 µg/mL streptomycin under standard culture conditions.
After 48 h of PTPN21 overexpression or knockdown in A549 cells, the cell culture media (CM) was collected and centrifuged at 800 g for 5 min at room temperature to remove cell debris. MRC-5 cells were digested and seeded at 5 × 104 cells per well in a 6-well plate with 2 mL of MEM complete medium. After 12 h, 1 mL of the original medium was discarded and replaced with 1 mL of the cell CM from A549 cells with PTPN21 overexpression or knockdown. The cells were then cultured for an additional 48 h.
Isolation of primary human lung fibroblasts and treatment
For detailed procedures, please refer to our previous publication [20]. Briefly, human lung tissue was dissected into small cubes and then digested in Hank’s balanced salt solution (Hyclone) containing collagenase (1 mg/mL; C0130; Sigma) at 37 °C for 30 min, followed by centrifugation at 500 g for 5 min. The tissue was then washed and resuspended in high-glucose Dulbecco’s modified Eagle medium (Hyclone) containing 10% fetal bovine serum, 100 U/mL penicillin, and 100 mg/L streptomycin (Solarbio). The tissue was incubated in a cell culture incubator at 37 °C with 5% CO2 for 30 min to allow the tissue fragments to settle. The medium was gently replaced to remove non-adherent cells and debris, and the culture was continued for 5 days to obtain primary human lung fibroblasts (PHLF). The subsequent treatment of PHLFs was the same as that for MRC-5 cells.
Reactive oxygen species assay
The ROS Assay kit (Solarbio, Beijing, China) was used to assay ROS levels in the A549 cells. The experimental procedures were according to the manufacturer’s instructions. Briefly, every well of 96-well plates was added 100 µL of DME/F-12 diluted DHE red fluorescent dye (5µM), and the cells were incubated in the dark at 37 ℃ for 30 min, then the staining solutions were removed and the cells were gently rinsed with warm HBSS with Calcium and Magnesium buffer for 3 times. Within 2 h of staining, the DHE red signaling were excited at 530 nm and read at 570 nm with a fluorescent multiplate reader or photographed under a fluorescence microscope.
To ensure that changes in ROS levels were not attributable to alterations in mitochondrial content, A549 cells were seeded in 96-well plates and subjected to identical treatments. Total DNA was extracted from the cells using a cell/tissue DNA extraction kit (Yeasen), following the manufacturer’s protocol. Mitochondrial DNA (mtDNA) and nuclear DNA were then amplified using the Hieff Canace PCR Master Mix (Yeasen) and the Human mtDNA Monitoring Primer Set (TaKaRa). The relative quantification of mtDNA copy number was determined according to the instructions provided. Finally, ROS fluorescence intensity was normalized by the relative mtDNA levels in the cells.
Mitochondrial superoxide assay
The MitoSOX™ Red (mitochondrial superoxide fluorescent probe) (Thermo Fisher, Shanghai, China) was used to detected mitochondrial superoxide. The experimental steps were followed by the manufacturer’s instructions. In short, after transfected the plasmids for 48 h, A549 cells in 96-plates were stained with 5 µM MitoSOX™ Red and incubated cells for 30 min at 37 ℃ and 5% CO2 in dark, then cells were washed gently 3 times with warm HBSS with Calcium and Magnesium buffer. The plates were excited at 518 nm and read at 570 nm using a fluorescent multiplate reader or photographed under a fluorescence microscope within 2 h of staining. Finally, the MitoSOX levels was normalized by the relative mtDNA levels in the cells.normalized to the relative mtDNA levels in the cells.
Mitochondrial membrane potential assay
The mitochondrial membrane potential (MMP) of the A549 cell in 96-well plates was measured by MMP assay kit with JC-1 (Beyotime, Shanghai, China). the cells were first washed with PBS, then 100 µL of cell culture medium was added to each well, and then equal volume of JC-1 staining working solution was added and thoroughly mixed. After incubating at 37 ℃ for 20 min, the supernatant was aspirated and washed with JC-1 staining buffer. The green and red fluorescence intensities of the cells were excited at 485 nm and 530 nm and read separately on a multifunctional enzyme plate. Next, calculate the red/green fluorescence ratio to evaluate changes of MMP.
Mitotracker staining
Mitotracker staining was used to visualize mitochondrial morphology in A549 cells. Sterile coverslips were placed in a 24-well plate, and A549 cells were seeded onto the coverslips. The cells were treated with either overexpression or knockdown of PTPN21. After 48 h, Mitotracker dye (Beyotime, Shanghai, China) was diluted in DF12 medium to a final concentration of 20 nM to prepare the working solution. A volume of 300 µL of this solution was added to each well to ensure complete coverage of the cells. The cells were incubated at 37 °C for 30 min, then washed three times with PBS to remove any excess dye. The cells were fixed with 4% paraformaldehyde for 5 min, followed by three additional PBS washes. The nuclei were stained with DAPI, and images were captured using a confocal microscope. Two random fields were imaged per group, and the number of fragmented mitochondria in each field was counted.
Detection of autophagic flow
The A549 cells were seeded on the coverslips in 24-well plates (3 × 104 cells/well). GFP-RFP-LC3 plasmid and pcDNA3.1-PTPN21 or control plasmid, and GFP-RFP-LC3 plasmid and pcDNA3.1-U6-shPTPN21 or the control plasmid were simultaneously transfected into cells using Lipofectamine 3000. After 48 h, the cells were treated with chloroquine (50 µM) for 5 h, then fixed with 4% paraformaldehyde and stained with DAPI working solution for 10 min. Images were obtained using a laser scanning confocal microscope (Leica, Wetzlar, Germany).
ATP level detection
ATP levels were measured using the ATP assay kit (Beyotime, Shanghai, China). The treated A549 cells were lysed on ice, then centrifugated at 12 000 ×g for 5 min, the supernatant (20 µl) is added to 100 µl of ATP assay working solution and mixed. The absorbance was measured with a multifunctional microplate reader. Finally, the obtained readings were normalized by the relative mtDNA levels in the cells.
Cell adhesion assay
After the plasmid was transfected for 48 h, the A549 cells were digested by 0.25% trypsin, and about 5 × 104 cells were added to one 24-well plates which precoated with human fibronectin (Med Chem Express, Shanghai, China) and three multiple wells were set in each group. The culture plates were incubated at 37 ℃ for 1 h, then non-adherent cells were removed by gentle washing three times with PBS, and the remaining cells were stained with 0.1% crystal violet for 5 min. After washing, images were taken under a phase contrast microscope on 100 × field.
Wound healing assay
After the plasmid was transfected for 48 h in 6-well plates and grown to 100% confluence, the cell monolayers were wound vertically with a steriled 200 µL pipette tip, and then gently washed with PBS. The culture medium was replaced with DME/F-12 containing 2% FBS. At 0 h, 12 h and 24 h, pictures were taken respectively under an inverted microscope to check the condition of wound closure.
Immunohistochemical (IHC)
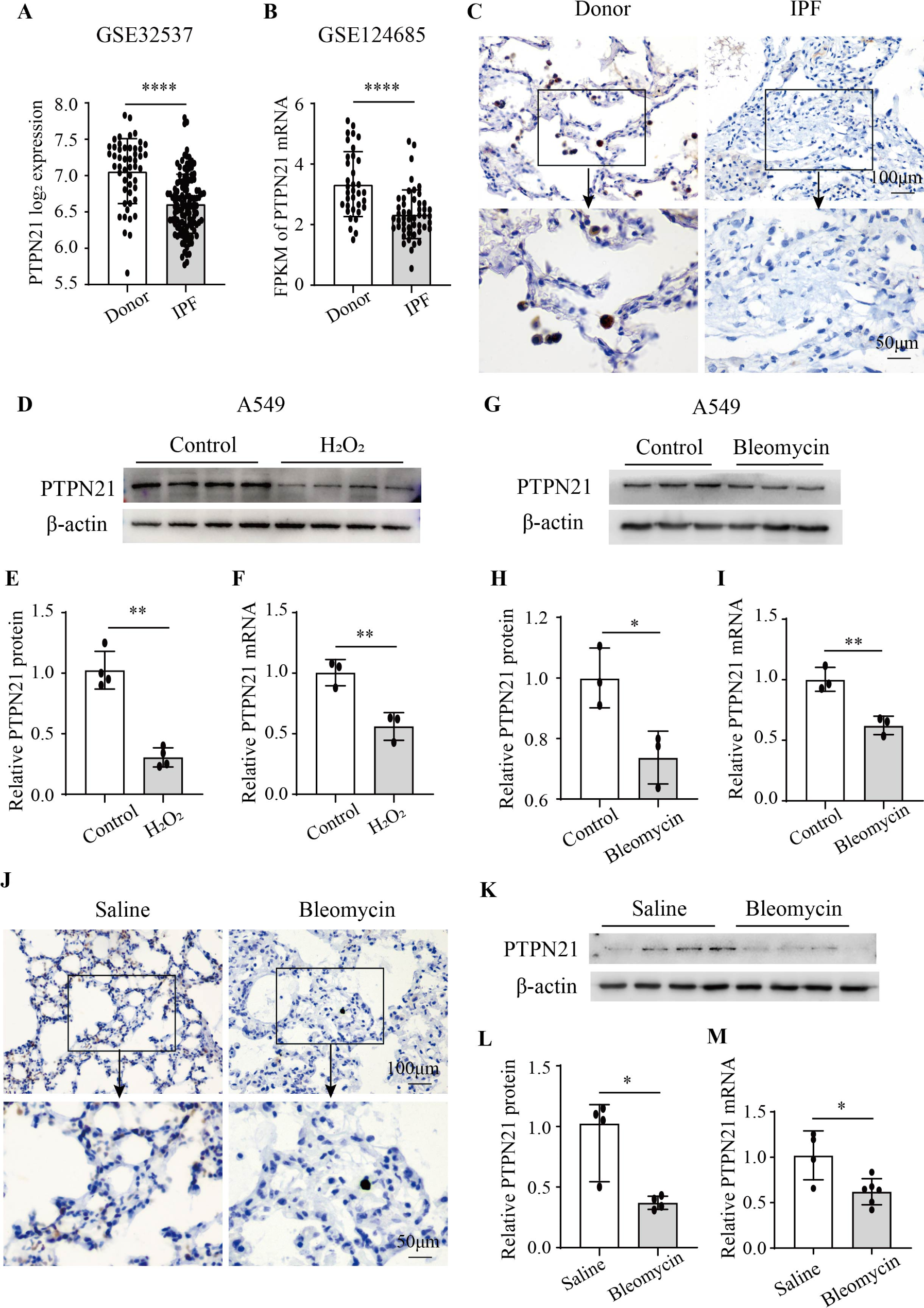
For IHC staining, mice or IPF paraffin-embedded tissue sections were successively deparaffinized in xylene, gradient ethanol, and subsequently added to PBS, then antigens of the tissue sections were repaired with sufficient sodium citrate buffer (pH 6.0) at 95 ℃ for 10 min and naturally lowered the temperature to room temperature. Next, the sections were permeabilized with 0.3% Triton X-100 and blocked with 5% goat serum at room temperature for 30 min, then incubated with the corresponding specific primary antibodies (PTPN21 (1: 100)) at 4 ℃ overnight. After washing with PBS, lung sections were incubated with biotin-labeled secondary antibodies (Beyotime, Shanghai, China) at 37 ℃ for 30 min. Then, a DAB working solution was used to develop the lung sections, followed by staining the nucleus with hematoxylin, the slices were sealed using neutral gum. The stained lung sections were photographed with a light microscopy. Lung tissue specimens from IPF patients were obtained from Henan Provincial Chest Hospital. This study was approved by the Medical Research Ethics Committee of Henan Provincial Chest Hospital (No. 2020-03-06).
Immunofluorescence (IF)
For the cell, the cells on the slides were fixed with 4% paraformaldehyde solution at room temperature for 10 min firstly, then neutralized with 2% glycine for 5 min, and softly washed with PBS. Before incubation with the second antibody, the procedures of IF of the lung tissue and the cells were the same as IHC. Lung tissue sections were incubated overnight at 4 °C with specific primary antibodies, including rabbit anti-PTPN21 (1:100, Abcam), mouse anti-Surfactant Protein C (SP-C) (1:100, US Biological), or mouse anti-EMR1(also named F4/80) (1:100, US Biological). After incubation, the sections were washed with PBS, the secondary antibodies of Alexa Fluor® 594/488-conjugated anti-rabbit IgG, and Alexa Fluor® 594/488-conjugated anti-mouse IgG (Cell Signaling Technology, Shanghai, China) were added and incubated at 37 ℃ for 1 h, then the nucleus was stained with a DAPI working solution. Cells were incubated overnight at 4 °C with a 1:100 dilution of the primary antibody α-SMA. The following day, a 1:400 dilution of phalloidin was added to the secondary antibodies, Alexa Fluor® 488-conjugated anti-mouse IgG, and incubated at 37 °C for 1 h, then the nucleus was stained with a DAPI working solution. Image acquisition used a Zeiss microscope or a confocal laser scanning microscope (Leica, WETZLAR, Germany).
Hematoxylin and Eosin (H&E) staining
H&E staining were carried out as described previously. Briefly, mouse paraffin-embedded lung tissue sections were routinely deparaffinized to distilled water. Then H&E staining were performed using the staining kit from Beyotime Biotechnology, following the manufacturer’s instructions.
Western blotting
Briefly, the total proteins of the mice lung tissues, A549 cells and MRC-5 cells were extracted, and the concentration of the proteins was determined as previously described. Then total proteins (30 µg) were run by sodium dodecyl sulfate-polyacrylamide gel electrophoresis gel and then transferred onto polyvinylidene fluoride membrane, followed by blocking the membrane with 5% fat-free milk for 45 min, the membranes were incubated with specific primary antibody (Table 2) (overnight at 4 ℃), then the corresponding horseradish-peroxidase-conjugated goat anti-mouse IgG or goat anti-rabbit IgG were added and incubated for about 1.5 h at room temperature. The immunoreactive bands were rinsed with a chemiluminescence reagent kit purchased from Beyotime Biotechnology, and images were taken by the image station (Gene Company Limited, Shanghai, China).
Table 2 The primary antibodies for Western blottingStatistical analyses
All data were statistically analyzed using GraphPad Prism 8 (GraphPad Software, Inc., San Diego, CA, USA). The Shapiro-Wilk test was used for normal distribution. The Mann-Whitney U test was used for comparison between groups with non-normal distribution, and the unpaired t test was used for comparison between groups with normal distribution. The data were expressed as mean ± standard deviation (SD), with statistical significance considered at p < 0.05.

留言 (0)