
記住我






This study aims to enroll coronary artery disease (CAD) patients with EA confirmed by IVUS at multiple centers, including Sir Run Run Shaw Hospital affiliated with Zhejiang University School of Medicine (lead unit), The First People’s Hospital of Hangzhou Lin An District, Zhejiang Greentown Cardiovascular Hospital, The Fifth People’s Hospital of Hangzhou Yuhang District and Ningbo Ninth Hospital.
Eligibility criteria Inclusion criteriaPatients’ inclusion criteria will be: (1) age ≥ 18 years; (2) CAD patients tolerating dual antiplatelet therapy, and clinically indicated for elective PCI; (3) IVUS confirms lesion length ≥ 20 mm and meets the characteristics of EA: plaque showing ultrasound signal attenuation (longitudinal attenuation length ≥ 5 mm), excluding calcified and dense fibrous plaques; (4) acceptance of terms and conditions of the study and signature of the informed consent form, with a full understanding of the trial content, process, and potential adverse reactions; (5) willing to adhere to medical follow-up.
Exclusion criteriaPatients’ exclusion criteria include (1) positive cardiac troponin I (cTnI) prior to percutaneous coronary angiography; (2) coronary artery bypass graft (CABG) lesion; (3) chronic total occlusion (CTO); (4) in-stent restenosis; (5) severe calcification; (6) left main coronary artery disease (LMCAD); (7) intervention of > 1 major coronary artery branch in a single PCI procedure; (8) side branch occlusion or blood flow-affecting dissection during PCI; (9) cardiogenic shock during PCI; (10) vulnerable populations, such as individuals with mental disorders, cognitive impairment, critical illness, pregnancy, lactation, genetic or acquired bleeding disorders with anticoagulation factor deficiency, or current use of oral anticoagulants.
Informed consentEach subject will be fully informed in writing about the purpose, nature, procedures, and potential benefits and risks of the study. Subjects should be informed that they have the right to withdraw from the study at any time. Before enrollment, each subject should be given sufficient time to consider participation. Only subjects who voluntarily sign the informed consent form after being fully informed will be included in the study. If a subject is unable to read the informed consent form (e.g., illiterate subject), a witness must observe the informed consent process and sign the informed consent form.
Who will take informed consent?Researchers will perform an initial eligibility screening and collect written informed consent. Written informed consent will be collected prior to baseline assessment.
Additional consent provisions for collection and use of participant data and biological specimensIf there is collection of any data that is not included in the initial informed consent process for the main clinical trial, each participant must sign an additional consent form for the ancillary study.
InterventionsExplanation for the choice of comparatorsIn this randomized controlled trial, participants will be randomly divided into the intervention group (intracoronary administration of 4 mL of 0.9% NaCl solution containing 4mg of TNK) and the control group (intracoronary administration of 4 mL of 0.9% NaCl solution). The control intervention allows the evaluation of TNK effects in preventing PCI-related MI. Patient education encompassing disease-related knowledge, lifestyle guidance, complication prevention, rational drug use, and other pertinent contents will also be delivered to the control group. Both the intervention group and the control group will receive standard pharmacological treatments.
Intervention description• Experimental group: Immediate intracoronary injection of 4 mL of 0.9% NaCl solution containing 4mg of TNK following balloon angioplasty (dilation).
• Control group: Immediate intracoronary injection of 4 mL of 0.9% NaCl solution following balloon angioplasty (dilation).
• The use of other devices during PCI will be decided based on the operator’s experience.
Criteria for discontinuing or modifying allocated interventionsDiscontinuation from the treatment program will be considered in the following conditions:
(1)Participants voluntarily withdraw from the study;
(2)If the participant is unable to continue the protocol treatment due to adverse events;
(3)The researcher(s) deem continued participation to be inappropriate or detrimental to the participant.
Strategies to improve adherence to interventionsRoutine follow-up is necessary for every patient who underwent PCI. Therefore, no extra measures will be taken to improve the adherence.
Relevant concomitant care permitted or prohibited during the trialBoth the intervention group and the control group will receive standard pharmacological treatments and hospital care.
Provisions for post-trial careThe administration of TNK aims to prevent the occurrence of PCI-related MI. The dosage of TNK used in our trial is reported safe in other studies. If any risk of harm is identified, the principal investigator will intervene to minimize the potential harm. Besides, the research team will provide appropriate compensation if necessary.
OutcomesOutcomes will be collected during hospitalization and again at 1-month follow-up after PCI.
Primary outcome measureIncidence of PCI-related MIPCI-related MI is defined as the increase of high-sensitivity cTnI to more than 5 times the 99th percentile upper reference limit (> 5 × 99th percentile URL) within 48 h post-PCI in patients with normal baseline cTnI values. Notably, PCI-related MI is a valid surrogate endpoint for subsequent MACE [1]. The rationale for employing a surrogate endpoint, rather than a clinical endpoint, includes (a) the significantly higher incidence rates of PMI compared to MACE in post-PCI conditions, which facilitates more straightforward and efficient observation of outcomes; and (b) the primary focus of this research on PMI, which is strongly associated with increased cardiac mortality and heightened risk of future MACE [1, 18] .
Secondary outcome measures 1.Proportion of patients with elevated postoperative high-sensitivity cTnI exceeding 5, 10, 35, and 70 times of the normal baseline.
2.Incidence of slow-flow after stent implantation and post-dilation.
3.Frame count of angiographic flow after stent implantation and post-dilation.
4.Incidence of MACE during hospitalization and post-operative 1-month follow-up.
Safety endpointsProportion of major bleeding (BARC bleeding classification type 2 or type 3) occurring within 24 h post-PMI.
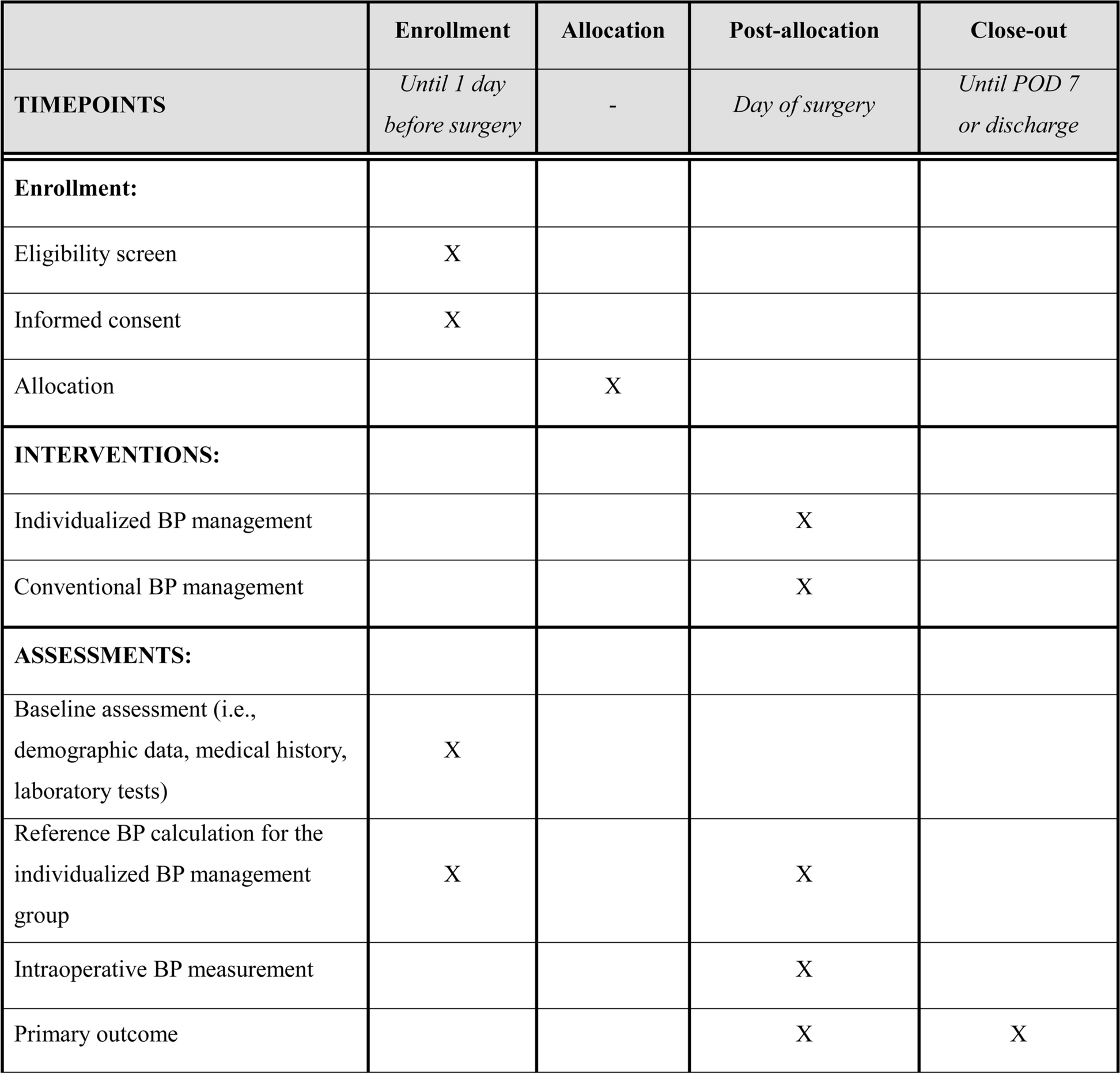
Participant timelineThe participant timeline is shown in Fig. 2.
Fig. 2 Sample size
Sample size This study is a RCT with an experimental group and a control group. The allocation ratio between the groups is 1:1. Based on previous literature and clinical data from our center, the assumed incidence rate of PCI-related MI in the control group is 20% [1, 3]. The experimental group is assumed to have a 35% lower incidence rate of PCI-related MI compared to the control group. Using a two-sided alpha of 0.05 and a power of 80%, a total sample size (N) of 720 patients was calculated using PASS 2023 software. Considering a 20% dropout rate due to loss to follow-up or refusal to follow-up, at least 864 subjects are required for the studies, with at least 432 subjects in each group.
RecruitmentParticipant recruitment will begin in August 2024 and is expected to continue until August 2025. We will recruit participants from the other four sub-centers, including The First People’s Hospital of Hangzhou Lin An District, Zhejiang Greentown Cardiovascular Hospital, The Fifth People’s Hospital of Hangzhou Yuhang District, and Ningbo Ninth Hospital.
留言 (0)