
記住我
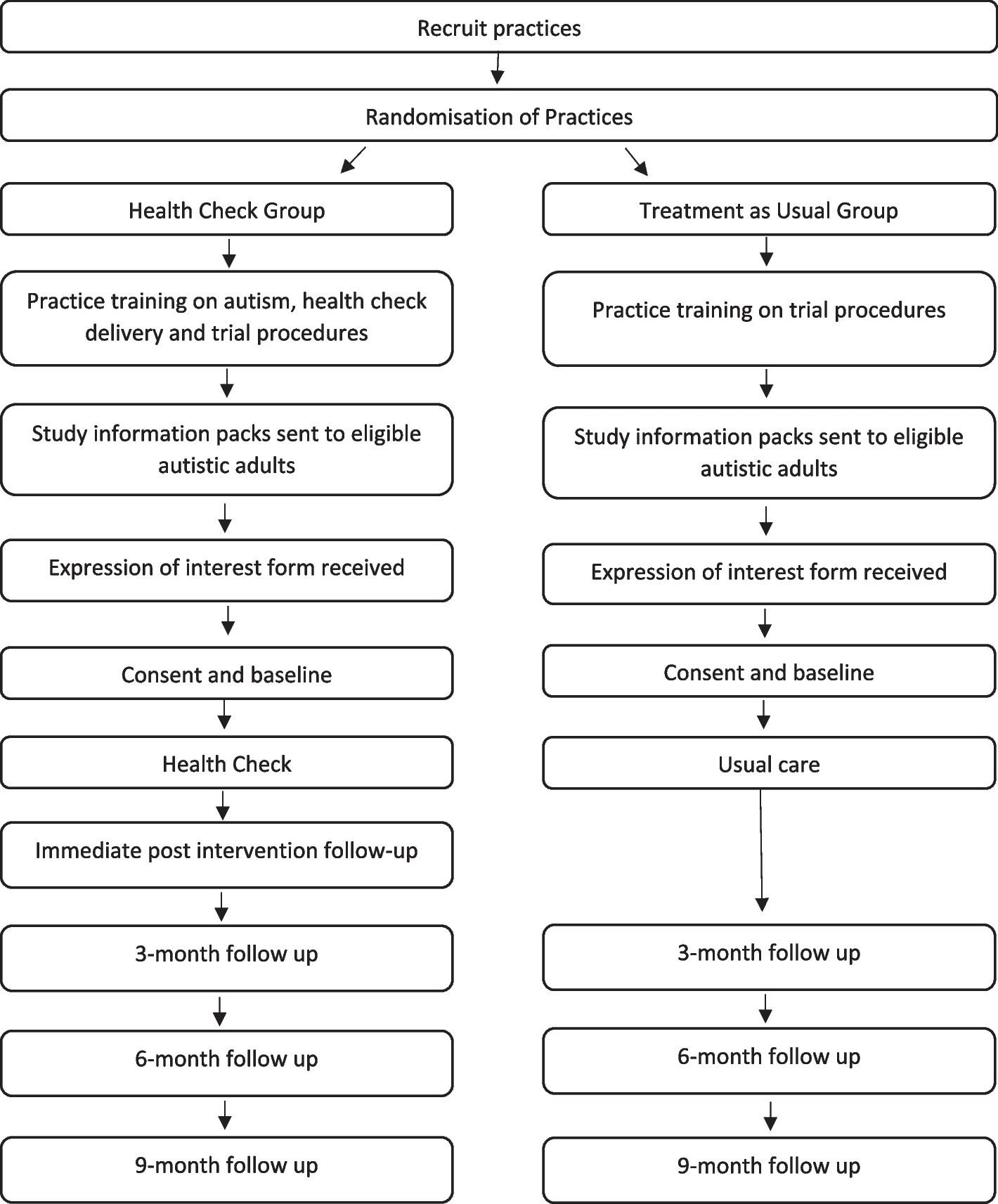






The present trial is designed as a prospective, double-blinded, triple-armed, multi-centered randomized controlled trial. The trial will be conducted following the guidelines Declaration of Helsinki, Standard Protocol Items: Recommendations for Interventional Trials (SPIRIT) [26], and Consolidated Standards of Reporting Trials (CONSORT) [27]. The trial design has been approved by the Institutional Ethics Review Board (IERB) (Letter No. D/137/FIMS) and is registered under the Clinical Trial Registry at www.clinicaltrials.gov with study ID NCT06155474. The flow diagram of the trail design is illustrated in Fig. 1.
Fig. 1 Study setting
Study settingThe trial will be conducted at two public sector District Health Quarter (DHQ) level hospitals in Lahore, i.e., Government Mian Mir DHQ and Government Samnabad DHQ. Permissions will be taken from the relevant authorities of the hospitals. These two hospitals are selected as currently Integrated Reproductive Maternal Newborn, Child Health (IRMNCH) & Nutrition Programs are running in these hospitals with a high patient load.
Study population and eligibility criteriaAll children suffering from SAM, without any complications, and aged 6–59 months, are eligible for inclusion in the present trial. Participants will be screened using weight-for-height/length z-score and mid-upper arm circumference (MUAC) as screening criteria. The criteria of the World Health Organization (WHO) will be used to diagnose edema among children. WHO defines children with grade 1–2 bilateral edema and a weight-for-height/length z-score of < − 3 standard deviations (SD) or MUAC of < 11.5 cm who are otherwise clinically healthy, aware, and with a decent appetite as children with SAM [28]. Trained healthcare staff including nurse, childcare worker, and doctor will assess the children for eligibility. MUAC will be assessed using WHO’s MUAC colored tapes in centimeters (cm), while weight-for-height/length z-score and weight of children will be assessed in grams (g) with minimal and light clothing using UNISCALE, and height will be measured using a standard measuring tape in cm(s) and then will be compared with the grading of z-score chart. All the anthropometric measures will be taken thrice to reduce information bias. Additionally, all the instruments that will be used for taking measurements will be checked for accuracy and zero precision beforehand. All eligible participants will be recruited following a pre-defined inclusion and exclusion criteria (Table 1).
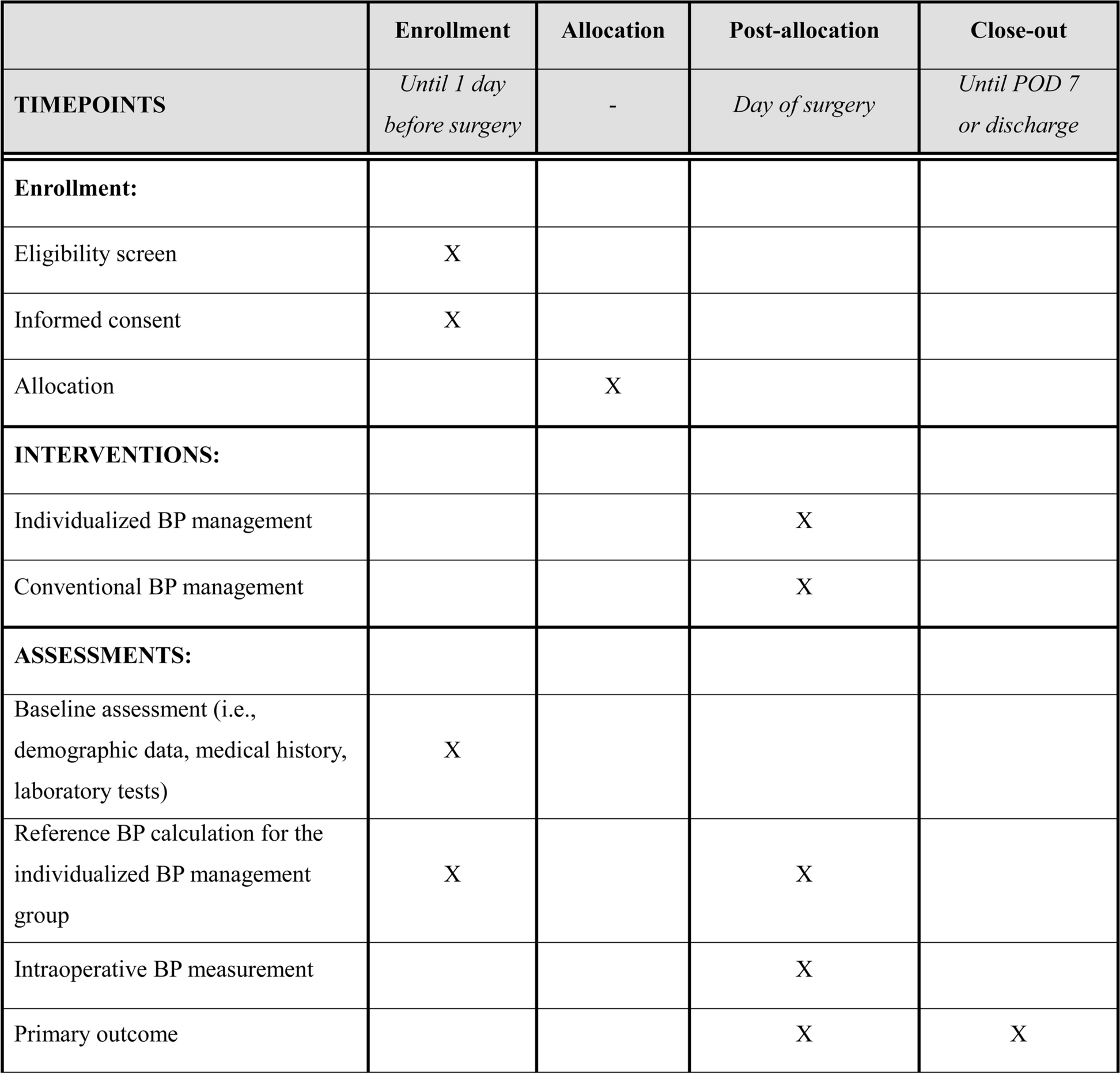
Table 1 Inclusion and exclusion criteriaSample size estimationFor sample size estimation, a balanced one-way analysis of variance power calculation with R software was used, and a minimum of sample of 53 participants was calculated for each group using 5% significance level, 80% power of study, 0.25 effect size (f) and 3 as k value (Fig. 2).
Fig. 2
Sample calculation using R software
A calculated sample of 159 participants (53 participants each arm) is further inflated by 10% to account for attrition due to loss to follow-up, making a total sample of 174 participants with 58 participants required to be recruited in each arm. For achieving adequate participant enrolment to reach target sample size, community mobilization in the nearby areas of the selected hospitals through lady health workers will be carried out with the support of IRMNCH & Nutrition Program.
Informed consentInformed consent will be taken from the parents/guardians of the children on a written consent form. The purpose of the trial and the potential adverse reactions (i.e., allergies) of the treatment modalities will be clearly communicated to them. Upon their approval, thumb impression will be taken on the written consent form. The process of taking consent from each parent/guardian will be conducted in the presence of at least one witness.
Randomization and allocationChildren suffering from acute malnutrition, otherwise healthy, aged 6–59 months, and fulfilling the inclusion criteria will be recruited randomly to one of the two arms of the present trial for 8 weeks, i.e., experimental and active comparator groups. However, healthy children with matched sex and age will be recruited from the vaccination centers of the research sites to the placebo comparator group as healthy controls. The participants in all groups will be recruited in equal allocation ratio (1:1:1) with no restriction of block size. A statistician unaware of the study method will generate the random numbers and allocate the participants to study groups by assigning unique code to them (i.e., SAM children to experimental and active comparator group and healthy children to placebo comparator group). All the participants will be given a recruitment card with their unique identity code mentioned on it.
BlindingA statistician unaware of the study, responsible for designating codes to the participants and allocation of groups, will prepare the packets of treatment regime for all the participants with day-wise dosage enclosed in separate opaque envelops and the unique code of the participants mentioned on it, blinding the participants and the investigator of the study. The treatment to all the participants will be given in separate rooms, and the parents of all the participants will be advised not to acquainted and communicate with each other after randomization for reducing the subjective biases. Blinding will only be permissible in case of withdrawal or termination of any study participants.
InterventionsThe detailed dietary intervention that will be provided to the participants recruited to each group is given in Table 2.
Table 2 Intervention for study groupsWithdrawal and termination criteria and managementRUTF will be provided as per the guidelines of WHO and all the therapies allowed by WHO will be permitted during the trial. However, children showing poor compliance or absence from two consecutive follow-up visits will be withdrawn from the study, while those showing any allergies or adverse events will be terminated from the study, and appropriate treatment will be provided to them after proper investigation by the principal investigator.
Adverse events and safety monitoringThroughout the trial, participants will be monitored on weekly basis to ensure safety. In case of any allergic reactions or adverse events, prognostic treatment will be provided after proper investigation free-of-cost. Further, participants will be encouraged to report to and contact the designated personnel in case of any discomfort or adverse event 24/7 to ensure the reporting and treatment of adverse events within 24 h. The principal investigator will then decide the continuation of the trial treatment after excluding for the factor unrelated to the trial medications. There are no anticipated harms of the interventions of the present trial; therefore, no post-trial care and compensation has been planned for participation in the present trial.
Strategies for improving adherenceFor improving the adherence of the trial, contact numbers of the parents of the participants will be collected, and a daily reminder text/call will be dropped. Empty treatment packets will not be collected for ensuring the blinding of the study. In addition to this, the parents of the participants will be incentivized through multi-vitamins for their other children and family members.
Outcome parametersAll the assessments will be carried out by a trained staff unrelated to the enrolment and allocation procedures. The primary outcome of the trial is to examine the microbiome diversity of malnourished children, to compare it with healthy children, and to evaluate the effect of RUTF plus prebiotics on the microbiome diversity of malnourished children (Table 3). For achieving the primary outcome, 5 g stool sample of the children will be collected for DNA extraction using QIAamp DNA Stool kit (Qiagen, Hilden, Germany) for examining the microbiome diversity through 16 Svedberg (S) ribosomal ribonucleic acid gene sequencing.
Table 3 Primary outcome parameterFor the accomplishment of secondary outcome parameters, anthropometric measures of the children, including their height, weight (for calculating weight-for-height/length z-score), and MUAC will be assessed by a trained healthcare staff. For assessing neurodevelopment of children, the Malawi Development Assessment Tool (MDAT) will be used that is considered to be one of the most valid tools for assessing child development for ages 0–59 months measuring their motor, language, social, and cognitive development [29], while for assessing the muscle mass accumulation, Body Stat Analyzer will be used (Table 4).
Table 4 Secondary outcome parametersData collectionAt baseline, sociodemographic characteristics of children including their age, gender, ethnicity, breast feeding history, mother’s demographics, mother’s nutritional status, and dietary habits will be recorded. The age of the child and the mother will be recorded through their national birth identity document, while the rest of the sociodemographic characteristics will be recorded through history of mother/parent/guardian. In addition to this, child’s anthropometric verbal measures (height, weight, and MUAC), neurodevelopment using MDAT, and muscle mass accumulation using body stat analyzer will be assessed, and 5 g stool sample of the children will be collected at baseline (week 0) and at the end of the trial (week 8) (Table 5). For collecting stool samples, stool samples kit will be used following the stool sample collection guidelines. Stool sample kits include a cardboard tube with inner metal container with one plastic container of preservation gel in it. For children using toilet seat, parents will be provided with a cardboard-and-tissue paper lining for fixing on the toilet seat, paper bowl for placing above the lining, disposable spoon for scooping the stool sample, and a sample collection tube for storing the sample. Parents will be instructed to ask the child to pass the urine before defecating the stool for ensuring that the sample is not contaminated with the urine and scoop out two spoons of the stool into the container. While for those children defecating in diapers, parents will be provided with one plastic wrap and will be instructed to place it in the diaper such that urine goes straight into diaper while the stool remains on the plastic wrap and to scoop out a sufficient amount into the container. All parents will be instructed to bring the sample the same day it has been collected. The samples will be labeled with the allocated unique identity codes and will be stored in a refrigerator at − 80 °C, while the rest of the data will be recorded using a structured performance by a trained healthcare staff. The timeline of the study is given in Table 5.
Statistical analysisIntention-to-treat (ITT) analysis will be used in the present trial for analyzing the results. All the participants who are randomized, received at least one intervention, and evaluated for at least one outcome will be analyzed with missing data substituted according to the principle of ITT.
For evaluating the microbiome diversity, Ribosomal Database Project (RDP) Bayesian algorithm will be used to identify microbial structure at phylum and genus level among samples and trial groups using Fast Length Adjustment of Short Reads (FLASH) version 1.2.11 and software R version 3.4.1.
Raw sequence data will be filtered to obtain high-quality clean data with DADA2, after which clean reads that overlapped each other will be merged into tags and clustered to operational taxonomic units (OTU). Taxonomic classifications will be assigned to OTU representative sequences using the RDP database. FLASH version 1.2.11 will be used for generating consensus sequences for paired-end reads, and software R version 3.4.1 will be used to create phylum and genus abundance bar plots. Further, alpha diversity analysis will also be conducted. Alpha diversity is the analysis of species diversity in a sample, measured by observed species index, Chao1 index, Shannon index, Simpson index, and good-coverage index.
Furthermore, Statistical Package for Social Science (SPSS) version 24 will be used for evaluating the change in weight-to-height/length z-score, MUAC, neurodevelopment assessment, and muscle mass accumulation among trial participants. Firstly, for descriptive analysis, percentages and frequencies will be presented of qualitative variables (gender of the child, ethnicity, breast feeding history, and mother’s dietary habits), while mean ± SD (in case of normally distributed variables) and median ± inter-quartile range (IQR) (in case of skewed variables) will be given for quantitative variables. Furthermore, the association of the sociodemographic characteristics of the children and mothers with the weight-to-height/length z-score, MUAC, neurodevelopment assessment, and muscle mass accumulation among children will be explored through regression analysis. For comparing the outcomes between three trial groups, test of analysis of variance (ANOVA) will be used with Scheffe and Games-Howell test for post hoc multiple comparisons in case of normally distributed data; otherwise, Kruskal–Wallis test will be applied, while for analyzing the data of repeated measures within the same groups, paired-sample t-test (for normally distributed datasets) or will Wilcoxon test (for non-normally distributed datasets) will be applied. All the tests will be applied at 95% confidence interval (CI) by an independent statistician blinded to the study.
Interim and additional analysesNo interim or temporary analysis has been planned for the present trial, as there are no anticipated harms of the interventions of the present trial.
Quality controlFor ensuring the quality of the data and trial, the trial steering committee, an Independent Data Monitoring Committee (IDMC) comprises a group of experts who are external to the clinical trial, and all the staff administered in the trial will be responsible. The administered trial staff responsible for randomization, allocation, data collection, and treatment administration will be provided with an extensive 2-day training by the principal investigator (first and second authors) before the initiation of the trial for introducing them to the specifics of the trial. Safety monitoring will be the responsibility of the Institutional Review Board (IRB) of University of the Punjab, who can inspect the records, documents, and assessors of the trial for ensuring the guiding principles of trial protocol with regular evaluation of data every alternate week during the trial. The IRB is independent of the study and does not have any competing interests. The trial steering group and the IDMC will conduct regular meetings every fortnight to review the conduct throughout the trial period for auditing.
Patient and public involvementNo patient and public were involved during the design of the present trial.
Protocol amendmentsAny changes to the protocol will be updated in the clinical trial registry. Afterwards, the principal investigator will notify the trial centers and a copy of the revised protocol will be added to the investigator site file. Any deviations from the protocol will be fully documented using a breach report form.
DisseminationThe results of the study will be published in a scientific journal, preferably open access, to reach the scientific community. In addition, the results will be communicated to the hospitals which are part of the study and, thereby, the health professionals working in these hospitals. There are no publication restrictions so that also the broader public will be addressed via postings in media.
Trial statusProtocol version: 1.3 of December 4, 2023.
Recruitment start (expected): June 15, 2024.
Recruitment finish (expected): July 31, 2024.
Trial registrationThe present trial is registered at ClinicalTrials.gov with identifier No: NCT06155474. Registration date is December 4, 2023.
留言 (0)