This study was conducted following the ethical guidelines approved by the Institutional Review Board of the University of Hong Kong/Hospital Authority Hong Kong West Cluster (IRB Ref. Number: UW 22-611) for collecting human peripheral blood mononucleated cells (PBMCs).
Cell culture
Human DPSCs (hDPSCs) were obtained from Lonza (Basel, Switzerland) and were cultured in α-modified Eagle's medium (α-MEM) (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% (v/v) fetal bovine serum (FBS) (Thermo Fisher Scientific) and 1% (v/v) P/S (penicillin/streptomycin) (Sigma-Aldrich, St. Louis, MO, USA). Cells between passages 3 to 6 were incubated in 5% CO2 at 37 °C for further experiments.
Time-dependent treatments in hDPSCs
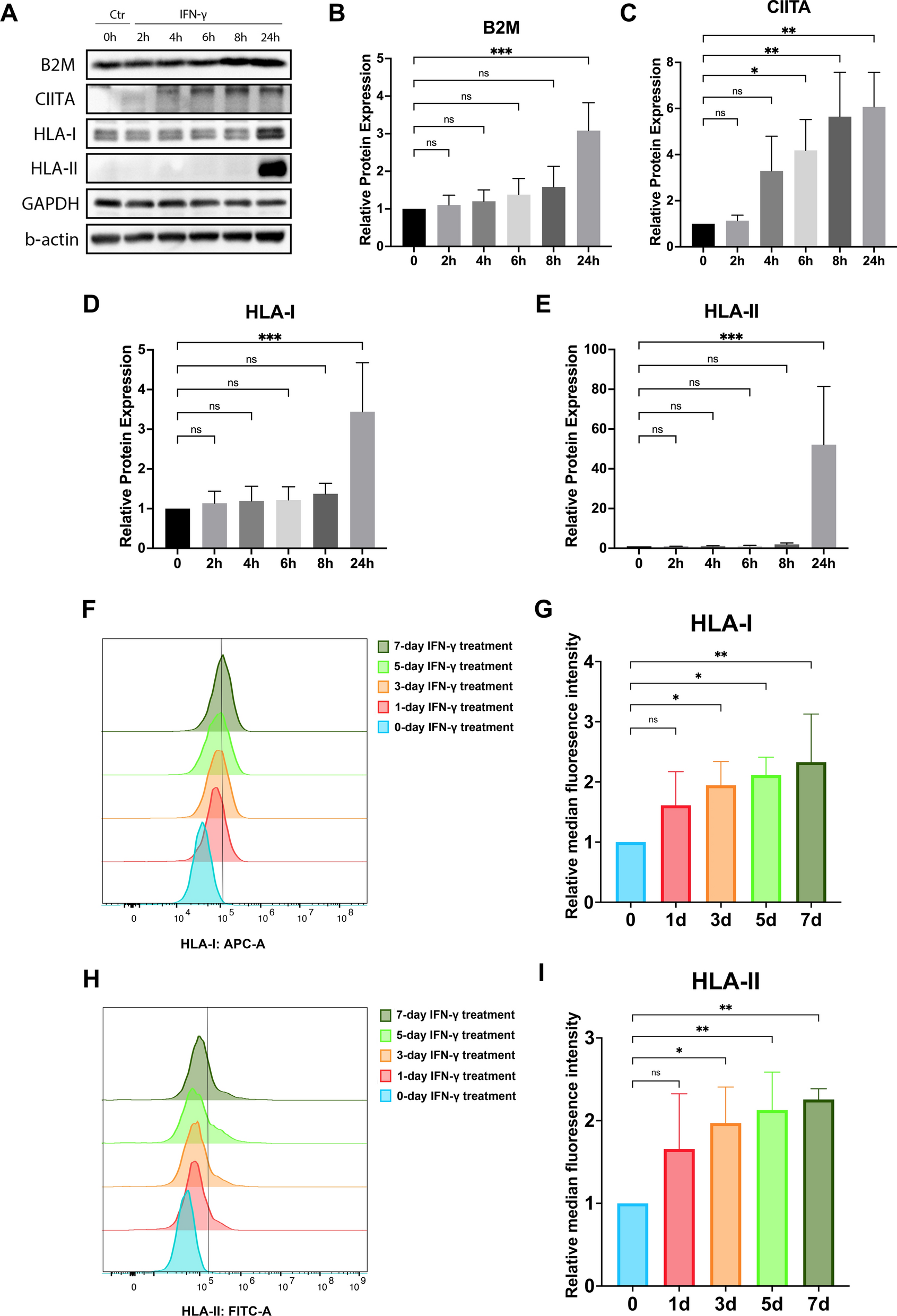
A series of time-dependent analyses were conducted on the target protein expression levels in hDPSCs after IFN-γ treatment to optimize the expression of B2M, CIITA, HLA-I, and HLA-II in subsequent experiments. The following were the details of the methodology:
To quantify the total protein expression of B2M, CIITA, HLA-I, and HLA-II in hDPSCs, cells were seeded in the 6-well plate at a density of 3 × 105 cells/well and treated with IFN-γ (10 ng/mL) in α-MEM supplemented with 10% (v/v) fetal bovine serum (FBS) and 1% (v/v) P/S (penicillin/streptomycin) under 5% CO2 at 37 °C. Samples were collected for Western blotting analysis after incubation at 0, 2, 4, 6, 8, and 24 h.
To track the localization of HLA-I and HLA-II in hDPSCs, cells were seeded in 12-well plate (1 × 105 cells/well) and cultured in α-MEM/10% FBS with or without IFN-γ (10 ng/mL) for 1, 3, 5, and 7 days; samples were then fixed with 100% iced methanal for immunofluorescence analysis.
To measure the cell surface protein expression of HLA-I and HLA-II in hDPSCs, cells were seeded in a 60-mm culture dish (Corning™, Corning, New York, USA) at a density of 3 × 105 cells/dish. They were treated in the α-MEM/10% FBS with or without IFN-γ (10 ng/mL). The medium was refreshed bi-daily. Samples were collected for flow cytometry analysis after 1, 3, 5, and 7 days.
Knockdown of B2M or CIITA in hDPSCs
B2M or CIITA knockdown was achieved using shRNA particles and the respective control-vector lentiviral particles (Santa Cruz Biotechnology, Texas, USA). Lentiviral transduction was performed as per the manufacturer's protocol. Briefly, hDPSCs were plated in a 6-well plate with 50% confluence. After attachment, Polybrene (Santa Cruz Biotechnology) at a concentration of 5 μg/mL was used to enhance infection efficiency, followed by adding 20 μL lentiviral particles (1 × 105 IFU) into the culture medium each well. Stably transduced hDPSCs (DPSCs-shCtr, DPSCs-shB2M, DPSCs-shCIITA) were selected using puromycin (Gibco™, Thermo Fisher Scientific) after 72 h. The knockdown efficiency was confirmed by Western blotting, Immunofluorescence, and flow cytometry, respectively.
Cell proliferation assay
The self-renewal capacity of DPSCs, DPSCs-shCtr, DPSCs-shB2M, and DPSCs-shCIITA was assessed by Cell Counting Kit-8 (CCK8, Dojindo, Kumamoto, Kyushu, Japan) following the manufacturer's instructions. Briefly, cells were seeded into a 96-well plate at a density of 7000 cells per well and incubated for 1, 3, 5, and 7 days at 37 °C with α-MEM/10% FBS refreshed every two days. On the assessment day, 10% CCK-8 solution was added and incubated for 3 h. Absorbance was measured at 450 nm using a spectrophotometer (SpectraMax M2, Molecular Devices, Sunnyvale, CA, USA).
Osteogenic differentiation
DPSCs, DPSCs-shCtr, DPSCs-shB2M, and DPSCs-shCIITA were seeded into a 12-well plate at 1 × 105 cells/well. To induce osteogenic differentiation, an osteogenic induction medium consisted of 10% FBS (Thermo Fisher Scientific), 10 Mm—glycerolphosphate (Sigma-Aldrich, Missouri, USA), 10 nM dexamethasone (Sigma-Aldrich), and 50 mg/L L-ascorbic acid (Sigma-Aldrich) was used to induce osteogenic differentiation. The medium was refreshed every 3 days. Cells were then fixed with 4% paraformaldehyde (PFA) for 30 min after a 21-day culture, and 2% Alizarin red (Sigma-Aldrich) was used for 10 min staining. The cells were photographed using a microscope (Nikon, Tokyo, Japan). The Alizarin Red staining area percentage was calculated using the Image J software (Bethesda, Maryland, USA).
Adipogenic differentiation
DPSCs, DPSCs-shCtr, DPSCs-shB2M, and DPSCs-shCIITA were seeded in a 12-well plate at 1 × 105 cells/well density. Then, the cells were cultured in the adipogenic induction medium (Cyagen Biosciences, Guangzhou, China) for 3 days followed by a 1-day adipogenic maintenance medium (Cyagen Biosciences). After a 21-day culture, cells were fixed with 4% PFA for 30 min and stained with Oil Red O solution (Sigma-Aldrich) for 30 min to identify lipid vacuoles. The Image J software (Bethesda) was applied to quantify the lipid droplet area.
Neurogenic differentiation
DPSCs, DPSCs-shCtr, DPSCs-shB2M, and DPSCs-shCIITA were seeded into a 12-well plate at a density of 1 × 105 cells/well and cultivated for 7 days in a neurogenic induction medium. The medium contained the components as follows: DMEM/F12: neurobasal [1:1] supplemented with 0.5% [v/v] N2, 1% [v/v] B27, 100 μM cyclic adenosine monophosphate (cAMP), 20 ng/mL basic fibroblast growth factor (bFGF), and 1% penicillin/streptomycin. Tuj1 was served as a neuron marker (#ab78078, Abcam, Cambridge, UK) [28]. All the chemicals were purchased from Thermo Fisher Scientific. The medium was refreshed every 3 days. The immunofluorescence analysis was done using the immunofluorescence method described below. The mean fluorescence intensity of Tuj1 after neurogenic differentiation was measured by Image J software (Bethesda).
Western blotting
Western blotting analysis for protein expression was performed as previously described [29]. Briefly, the protein extracts were loaded in equal amounts, separated by SDS-PAGE, and then transferred to PVDF membranes. The PVDF membranes were then blocked in 5% milk at room temperature for 1 h and incubated with the primary antibodies listed below (diluted according to manufacturers’ instructions) overnight at 4 °C: anti-B2M (#ab75853, Abcam), anti-CIITA (#sc-13556, Santa Cruz Biotechnology), anti-HLA-ABC (#ab225636, Abcam), anti-HLA-DR (#sc-53319, Santa Cruz Biotechnology), anti-β-actin (#sc-47778, Santa Cruz Biotechnology), and anti-GAPDH (#2118S, Cell Signaling Technology, MA, USA). Following three washes, the secondary antibody conjugated with HRP was used for membrane incubation and then detected by WesternBright ECL HRP substrate (Advansta, San Jose, CA, USA). Image J software (Bethesda) was used for quantification.
Immunofluorescence
Cells were fixed with either 4% (w/v) cold PFA for 15 min followed by 0.3% (v/v) Triton X-100 in PBS containing 5% FBS permeabilization for 1 h or 100% cold methanol for 5 min followed by PBS containing 5% FBS blocking for 1 h. Cells were then incubated with primary antibodies against HLA-ABC (#ab225636, Abcam), HLA-DR (#sc-53319, Santa Cruz Biotechnology), or Tuj1 overnight at 4 °C. The secondary antibodies employed were Alexa Fluor 488®-conjugated goat anti-mouse antibody and Alexa Fluor 647®-conjugated goat anti-rabbit antibody (Cell Signaling Technology). The nuclei were stained with DAPI (Thermo Fisher Scientific). Images were taken using a fluorescent microscope (Nikon) and quantified by Image J software (Bethesda).
Flow cytometry
DPSCs-shCtr, DPSCs-shB2M, and DPSCs-shCIITA were collected after treatment and resuspended to approximately 1 mL iced flow cytometry buffer containing 1% PBS, 10% FBS (Thermo Fisher Scientific), and 0.1% sodium azide (S2002-100G, Sigma-Aldrich). Cells were then labelled with primary antibody for 30 min at room temperature in the dark. The anti-HLA-ABC (#ab225636, Abcam) and anti-HLA-DR (#sc-53319, Santa Cruz Biotechnology) were diluted according to the manufacturer's instructions. To remove unbound antibodies, cells were washed with PBS by centrifugation at 1000 rpm for 5 min. Alexa Fluor 488®-conjugated goat anti-mouse antibody and Alexa Fluor 647®-conjugated goat anti-rabbit antibody (Thermo Fisher Scientific) were then added to the cell suspension at the optimal dilution and allowed to incubate for at least 20–30 min at room temperature. The stained cells were analysed by The NovoCyte Advanteon BVR analyser (Agilent Technologies, Santa Clara, California, USA) and FlowJo Software (TreeStar, Ashland, OR, USA).
Mixed leukocyte-mediated cytotoxicity
To investigate the immunogenicity of DPSCs-shCtr, DPSCs-shB2M, and DPSCs-shCIITA, the leukocyte-mediated cytotoxicity was evaluated after the DPSC and PBMC coculture. Fresh PBMCs were separated from healthy volunteers using density gradient centrifugation with Ficoll-Hypaque (Invitrogen, Thermo Fisher Scientific), the Informed Consents were obtained from all the volunteers before the blood drawing. PBMCs were then resuspended in a culture medium (RPMI 1640, Thermo Fisher Scientific) containing 10% FBS, 50 μmol/l β-mercaptoethanol, nonessential amino acids, and l-glutamine. The 3-day IFN-γ pre-primed DPSCs-shCtr, DPSCs-shB2M, and DPSCs-shCIITA were seeded in a 96-well plate at a density of 1 × 104 cells/well. On the day of the experiment, allogeneic PBMCs were cocultured with DPSCs at a ratio of 12.5:1 in RPMI 1640/10% FBS for 3 days. DPSCs and PBMCs cultured alone were set as the control groups. After coculture, the leukocyte-containing culture medium was collected and centrifuged at 400 × g for 15 min. According to the manufacturer's instructions, the supernatants were incubated with the reaction mixture from the CyQUANTTM LDH Cytotoxicity Assay Kit (ThermoFisher Scientific). The lactate dehydrogenase (LDH) released from damaged cells was proportional to optical density measured at 490 nm with a reference filter of 680 nm. To rule out biased results due to LDH released from damaged PBMCs during coculture, CCK-8 was applied to evaluate the survival of adherent gene-modified cells after removing suspended cells.
T lymphocyte proliferation assay
Human T cells were negatively isolated from PBMCs using the Pan T cell kit (Miltenyi Biotech, Bergisch Gladbach, Germany) per the manufacturer's instructions. The isolated T cells were assigned to the following experiments:
To evaluate the proliferation of Pan T cells following coculture, isolated T cells were stained with prewarmed 10 μmol/L Vybrant CFDA SE (CFSE)/phosphate-buffered saline staining solution (Invitrogen, Thermo Fisher Scientific) according to the manufacturer's instructions. T cell activation was achieved by mixing the CFSE labelling T cells with ant-CD3/anti-CD28 microbeads (Dynabeads™ Human T-Activator CD3/CD28, Thermo Fisher Scientific) in a 1:1 ratio in RPMI 1640/10% FBS medium. The activated T cells were then cocultured with 3-day IFN-γ pre-primed DPSCs-shCtr, DPSCs-shB2M and DPSCs-shCIITA in the RPMI 1640 medium supplement with 10% FBS, 50 μmol/L β-mercaptoethanol, nonessential amino acids, l-glutamine, and interleukin-2 (IL-2, 30 U/mL, BD Biosciences, San Diego, CA, USA) and incubated in a humidified incubator at 37 °C. After a 4-day co-culture, the T cells were collected for flow cytometry analysis after removing all the microbeads. The control groups were crucial in this experiment: gene-modified DPSCs only, activated CFSE-labelled T cells only, and T cells cultured in the absence of CFSE staining and CD3/CD28 activation. The activated CFDA-labelled T lymphocytes were monitored by flow cytometry after the coculture. The non-proliferating population was determined based on the negative control peak (T cells cultured alone). The quantification of T cell proliferation was reported as proliferating T cell percentage and proliferation index (PI). The PI is calculated by dividing the total number of cell divisions by the number of cells that underwent division. This index specifically accounts for responding cells, meaning it only includes cells that have undergone at least one division. It would be prudent to evaluate this value as it exclusively considers the percentage of cells that have been responsive.
To investigate the CD4 and CD8 expression in Pan T cells after co-culture, isolated T cells were activated by anti-CD3/anti-CD28 microbeads before being co-cultured with gene-modified DPSCs in an incubator for 4 days. After collecting the T cells, the Fc receptor antibody (BD Biosciences) was added to block the T cells for 15 min before a 30-min primary antibody incubation. The primary antibodies were diluted according to the manufacturer's instructions: anti-CD4 (#ab213215, Abcam) and anti-CD8 (#ab17147, Abcam). The secondary antibodies were applied after PBS washing. Flow cytometry analysis was used for CD4 and CD8 expression quantification.
Statistical analysis
All experiments were completed in triplicates independently, and all data were presented as mean ± standard deviations (SD). Statistical analysis was performed using the GraphPad Prism software version 6.0 (IBM, Armonk, NY, USA). One-way ANOVA with Dunnett's post hoc test or Two-way ANOVA with Bonferroni's post hoc test were used in multiple comparisons. A p value ≤ 0.05 was considered statistically significant.






留言 (0)