
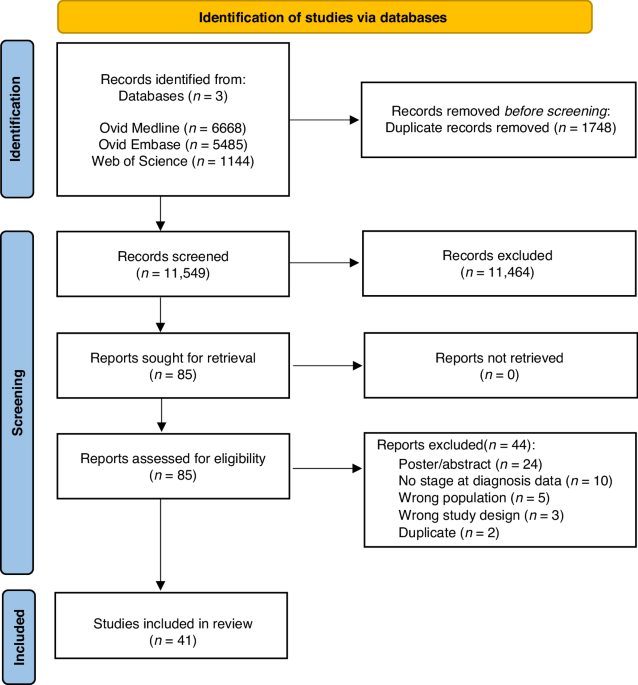
記住我
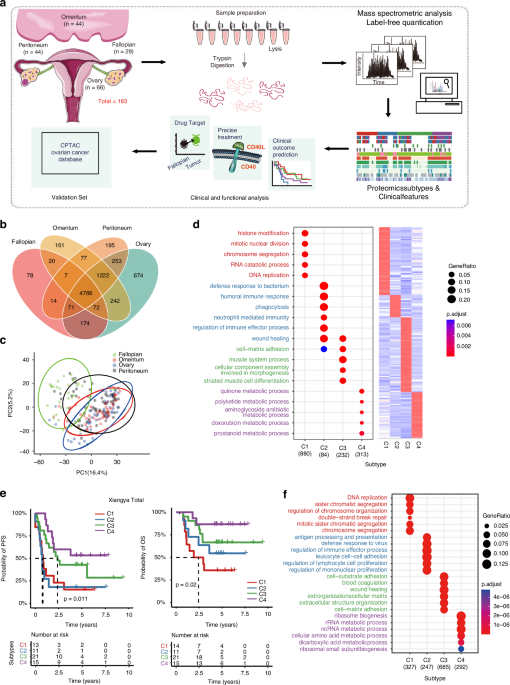
To comprehensively investigate the EOCs covering various pathological statuses, we prospectively collected 183 specimens, including 154 EOC tumour samples and 29 normal fallopian tube tissues. All specimens were obtained from 82 patients before undergoing any treatment. The tumour samples, including 22 primary tumour lesions, 44 primary tumour lesions and their paired omental and peritoneal metastatic lesions) (Fig. S1a, b, detailed sample collection can be found in STAR Methods and Table S1a). All those tumours were acquired through laparoscopy or primary debulking surgeries [16], covering a diverse range of pathological and histological statuses of epithelial ovarian cancer (EOC), including 137 high-grade serous carcinomas (HGSCs) and 17 samples of other histological subtypes (Fig. S1a, b, Table S1a, see Method details). Label-free liquid chromatography (LC)–tandem mass spectrometry (MS/MS) with data-dependent acquisition was conducted, and quantification was performed using iBAQ followed by normalization to iFOT [17,18,19]. Nonnegative matrix factorization (NMF) analyses [20] were carried out on the 183 specimens for the classification of proteomic subtypes (Fig. 1a). Comprehensive details regarding sample information and clinical features are summarized in Table S1a. The subtyping results were then integrated with clinical information to elucidate the relationships between molecular characteristics and therapeutic outcomes (Fig. 1a).
Fig. 1: Integrated OC proteomic analysis identifies four Proteomic subtypes associated with prognosis.
a Workflow diagram for sample collection, MS-based quantitative proteomics, bioinformatic analyses and validations. Following lysis, protein purification, and tryptic digest, peptides were separated by ultra-high performance liquid chromatography and measured in single runs using a quadrupole Orbitrap mass spectrometer. Label-free proteome quantification was performed using the MaxQuant software environment. b Venn diagrams depicting overlapped proteins detected in the primary ovary, omentum, peritoneum tumour sites and fallopian tubes. c PCA analysis of samples from tissue sites as indicated in Xiangya OC proteomics data. d The proteomic average abundance of signature genes for 4 NMF subtyping (right) and the dot plot showing GO-BP (biological progress) analysis of feature proteins referring to Xiangya four Proteomic subtypes (left). e The Kaplan-Meier (K-M) curves of progression-free survival (PFS, left) and overall survival (OS, right) for Proteomic subtypes for Xiangya OC patients. The tables below show the numbers of patients in follow-up at the year as indicated. f Dotplot showing GO-BP (biological progress) analysis of feature proteins for Proteomic subtypes of 2023 OC validation cohort.
Overall, the proteomics analysis detected 8032 gene products as reliable identifications, which were used for further analysis (Fig. S1c, Table S2a, See STAR Methods). The number of proteins detected in each sample ranged from 1382 to 4268; more than 2500 proteins were detected in over 85% of samples (Fig. S1d), and 4766 proteins were detected in all samples (Fig. 1b). The number of proteins detected at the tumour sites was higher than that detected in the fallopian tubes. The relative abundance (iFOT) of the proteins after logarithmic transformation showed a distribution of approximately ten orders of magnitude (Fig. S1d). Principal component analysis (PCA) revealed well-separated distributions between fallopian tubes and tumour sites. In contrast, the three types of tumour sites exhibited no discernable differences, indicating similarity in tumour proteomes regardless of primary or metastatic sites (Fig. 1c).
To conduct proteomic subtyping, we first selected the top 2500 most abundant proteins detected in each sample, yielding a total of 6,424 proteins (Table S2B). We then selected proteins that were detected in more than ten percent of all samples (18/183 samples) with a coefficient of variation (CV) of abundance greater than 2, resulting in 1044 proteins (Fig. S1e, Table S2c). We scored the 1044 protein expression signatures and employed NMF consensus-clustering for subtyping (Fig. 1d, Fig. S1f, g, h). The NMF clustering yielded four subgroups with a maximum average silhouette of 0.78, namely, cluster 1 (C1, n = 40, 22%), cluster 2 (C2, n = 57, 31%), cluster 3 (C3, n = 48, 26%) and cluster 4 (C4, n = 38, 21%) (Fig. S1e, F). Gene Ontology (GO) [21] enrichment analyses further elucidated that C1 showed cell proliferation gene signatures (Fig. 1d, Table S2d). C2-enriched proteins were involved in immune response pathways; the C2 subtype also had elevated expression of chemotaxis proteins including ITGB2 and SERPINE1, which are required for immune cell migration [22,23,24]. C3-enriched proteins showed high association with cell-matrix adhesion and muscle system processes, such as LAMA5, TNXB and several collagen proteins. C4-enriched proteins were mainly involved in various metabolic pathways (Fig. 1d, Table S2d).
We assessed the correlation between proteomic subtypes of primary tumours and patient prognoses. Patients in C3 and C4 showed better prognoses, while those in C1 and C2 exhibited less favourable outcomes, as evidenced by the progression-free survival (PFS) and overall survival (OS) curves (Fig. 1e). Utilizing a similar subtyping procedure, the CPTAC and 2023 Ovarian Cancer (OC) validation cohorts can also be divided into 4 distinct proteomic subtypes with high average silhouette width (0.79, Fig. S1i, j, for CPTAC cohort; 0.78, Fig. S1l, m for 2023 OC cohort) and consistent molecular characteristics (Fig. 1f, Fig. S1k, Table S2e, f), further indicating the reliability of subtyping and the consistency in molecular characteristics. Intriguingly, the association of PFS with proteomic subtype was also observed in HGSC patients (Fig. S1n).
The clinical relevance of the four proteomic subtypesTo assess whether the proteomic subtype could serve as an independent prognostic factor, we performed multivariate Cox regression analysis by controlling clinical characteristics. These included established factors such as age, residual tumour at surgery, timing of surgery, and FIGO stage, along with factors exhibiting prognostic correlation in univariate Cox analysis in our cohort (HE4, CA125). The results demonstrated that the proteomic subtype served as a robust and independent prognostic biomarker for evaluating OC patient OS (Table 1) and PFS (Table S1c).
Table 1 OS Multivariable Cox Regression Analysis (95% CI) for XY OC.To further validate the clinical relevance, we analysed the correlations of clinical events with the four proteomic subtypes (Fig. 2a, b, Table S1a). Notably, among the four subtypes, patients with early-stage disease (FIGO stages I and II) and no distant metastasis predominantly clustered within the C3 subtype, suggesting a more favourable prognosis for this group (Fig. 2b). Intriguingly, 79.3% of fallopian tube samples were classified as C3 (Fig. 2a, b), suggesting a molecular relationship between the fallopian tube and C3 ovarian cancer.
Fig. 2: The four Proteomic subtypes show distinct clinical relevance.
a The association of proteomic subtypes with 7 clinical variables. Kruskal-Wallis test was used for continuous variable CA125 (U/ml, the serum CA125 before debulking surgery or neoadjuvant chemotherapy). Fisher’s exact tests were used for other categorical variables (***p < 0.001, **p < 0.01, *p < 0.05). (Time of Surgery: PDS = primary debulking surgery, IDS = interval debulking surgery, refers to surgery after neoadjuvant chemotherapy (NACT)). b The distribution of samples with 4 Proteomic subtypes in different FIGO stages, metastasis, and tissue sites. c The distribution of samples with different platinum response in 4 Proteomic subtypes. d The distribution of serum CA125 level in 4 Proteomic subtypes. (Kruskal-Wallis test, **p < 0.01, *p < 0.05).
Most patients with FIGO stage IV disease were concentrated within the C1 and C2 subtypes (83.3%, Fig. 2b). Patients experiencing relapse within 6 months, between 6 to 12 months, and beyond 12 months after completing prior platinum-based chemotherapy were categorized as platinum-resistant, partially platinum-sensitive, and platinum-sensitive, respectively (detailed definition see Additional Clinical Data). The C1 and C2 subtypes exhibited a higher proportion of patients with reduced chemotherapy response, including both platinum-resistant and partially platinum-sensitive cases (C1: 57.1% [8/14]; C2: 61.5% [8/13]; C3: 22.7% [5/22]; C4: 5.8% [1/17]) (Fig. 2c). Additionally, patients in the C1 subtype showed elevated serum CA125 levels (Fig. 2d). These correlations indicate a poorer prognosis for patients in the C1 and C2 subtypes. In contrast, the C4 subtype was associated with a favourable response to platinum-based therapy (Fig. 2c, 83% of C4 cases were platinum-sensitive) and had the second-highest proportion of early-stage patients (Fig. 2B), further supporting its link to the best prognosis. However, no strong relationship between the proteomic subtypes and other factors, including surgery, HE4 and ascites, was observed (Fig. S2a, b). Thus, we show that the four proteomic molecular subtypes exhibit distinct molecular features and clinical relevance.
ScRNA-seq reveals the cell ecosystem of each proteomic subtypeTo better understand the molecular details of the four proteomic subtypes, we collected an additional 8 fresh primary tumours and performed scRNA-seq to study the cell ecosystem (Table S1b). We first performed proteomic profiling on the 8 samples (7 HGSCs and 1 clear cell carcinoma). NMF classified the 8 samples into C1 (1 sample, HGSCs), C2 (4 samples, 4 HGSCs), C3 (1 sample, 1 HGSC) and C4 (2 samples, 1 HGSC and 1 clear cell carcinoma) (Fig. 3a and Fig. S3a). High-quality transcriptomes of 40,710 single cells [25] were obtained based on the 10X Genomics platform (Table S3a). Using graph-based clustering, we first stratified them into 6 major cell populations: epithelial cells, endothelial cells, fibroblasts, T cells (including natural killer (NK) cells), B cells, and myeloid cells (including mast cells, macrophages, dendritic cells (DCs), and monocytes) (Fig. 3b). The composition and expression levels of key cell type markers confirmed the assignments for the cell clusters (PECAM1 and CDH5 for endothelial cells, EPCAM, KRT8 and CDH1 for epithelial cells, DCN, COL1A1, COL1A2 and PDGFRA for fibroblasts, CD3D and CD3E for T cells, CD79A, CD79B, CD19 and MS4A1 for B cells and LYZ, CD14, CD163 and CSF1R for myeloid cells) [26,27,28] (Fig. 3c, d). The labelling of the epithelial cell clusters was further supported by high copy number variation (CNV) scores (Fig. S3b).
Fig. 3: scRNA-seq analysis revealed the cell ecosystem of each proteomic subtype.
a Uniform manifold approximation and projection (UMAP) showing the distribution of 8 scRNA-seq samples. b UMAP plot showing 6 cell clusters for 8 samples analysed by 10X scRNA-seq (Left). Ratio of 6 cell types relative to the total cell count per sample (Right, the total cell count was scaled to 1). c Bubble plot showing marker genes across 6 cell clusters. Dot size indicates fraction of expressing cells, coloured according to z-score scaled to expression levels. d UMAP plot showing expression levels of selected genes. e Heatmap showing the expression signature of top marker genes for 6 cell clusters. Expression signature of differential cell type specific proteins among 4 Proteomic subtypes from Xiangya OC proteomics (f), CPTAC 2016 OC cohort (g) and 2023 OC cohort (h). Colour of each cell indicates Z score (Log2 of global protein abundance scaled to proteomic expression standard deviations) of the protein in each sample. Annotations indicate 6 cell clusters (left) and Proteomic or mRNA subtypes or gene mutation status (up).
The genes that were upregulated in each cell type (see STAR Methods) were considered as their distinctive signature genes (Fig. 3e, Table S3b). Subsequently, we assessed these cell type signature genes in the 183 original EOC samples, characterizing their cell types based on their protein expression levels. As shown in Fig. 3f and Table S4a, upregulated genes in C1 and C4 clusters exhibited similar cellular characteristics, predominantly enriched in epithelial-associated genes. C2 encompassed numerous immune cell-related genes, in consistency with the scRNA-seq analysis which revealed a high abundance of immune cells in C2 subtype samples (Fig. S3c). In contrast, C3 predominantly featured fibroblasts and endothelial genes (Fig. 3f). Similar gene signatures of cell clusters were also observed in the four subtypes in the CPTAC 2016 validation cohort and 2023 OC cohort (Fig. 3g, h, Table S4b). While we did not observe a clear correlation between BRCA mutations and the subtypes in CPTAC 2016 cohort. Based on cell type distribution and the molecular signatures (Fig. 1), we designated C1 (with the worst prognosis and highly expressed epithelial cell-related genes) as the malignant proliferative subtype, C2 (with the highest proportion of immune cells and highly expressed immune cell and cell-killing related genes) as immune infiltrating subtype, C3 (encompassing approximately 80% of the fallopian tubes and exhibiting high expression of fibroblast and endothelial signature genes) as the Fallopian-like subtype, and C4 (displaying a high percentage of epithelial-associated genes and enrichment in genes related to mitochondrial respiration) as the differentiated subtype.
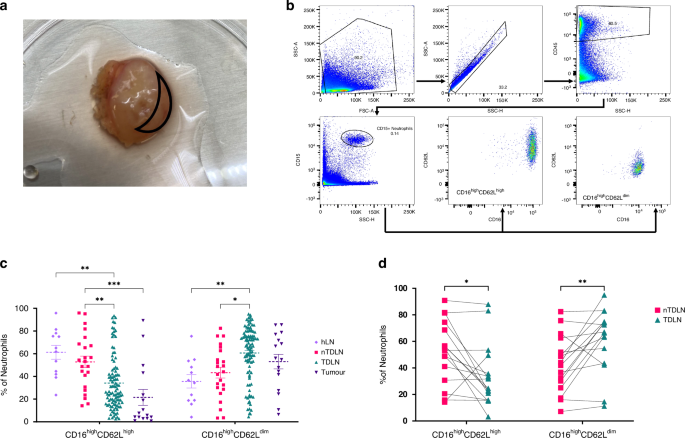
The immune infiltrating subtype is enriched with GZMK CD8+ T cells and MRC1 TAM-like macrophagesConsistent with the gene and protein signatures, the immune infiltrating C2 subtype contained a high percentage of immune cell infiltration (Fig. S4a), while the clinical outcome is worse in our proteomic analyses. We next stratified the 8 scRNA-seq samples into 4 immune (C2) and 4 non-immune groups (C1, C3 and C4) according to our proteomic subtypes, and analysed the immune cell subpopulations. The immune infiltrating subtype contained higher percentages of CD45+ immune cells (Fig. S4b–d), T cells and CD14+ myeloid cells (Fig. 4a, b). Fluorescence-activated cell sorting (FACS) analysis further verified the immune cell percentages. As shown in Fig. 4c, CD45+ cells, particularly T cells, were indeed enriched in immune infiltrating subtype tumours, consistent with the scRNA-seq results. B-cell and myeloid cell populations showed small and statistically insignificantly higher percentages in C2 tumours (Fig. 4c, Fig. S4e).
Fig. 4: The immune infiltrating subtype (C2) is enriched with highly infiltrative GZMK CD8+ T cells and MRC1 TAM-like macrophages.
a UMAP plot showing the distribution of 6 cell clusters in C2 (left, immune infiltrating subtype) and C1/C3/C4 proteomic subtypes (right). b Barplot showing percentage of 6 cell subpopulations in C2 and C1/C3/C4 subtypes. (*p < 0.05, error bar indicates ±SEM, two-sided Wilcoxon test) c Barplot showing the flow cytometry analysis of CD45+ cells (left), T cells (CD45+ CD3+), b cells (CD45+ CD19+) and Macrophage cells (CD45+ CD11b+) in C2 subtype compared with and C1/C3/C4 subtype. (*p < 0.05, error bar indicates ±SEM, two-sided Wilcoxon test) d UMAP plot showing 7 T cells subclusters from 4 C2 subtype patients. e Barplot showing percentage of 7 T cell subclusters in C2 subtype. f Bubble heatmap showing the expression of immune checkpoint genes across 7 T cell subclusters. G UMAP plot showing myeloid cells from 4 C2 patients. h Violin plot showing the expression of selected marker genes across 7 myeloid cell clusters. i Barplot showing the percentage of myeloid cells subclusters in C2 subtype. J Bubble heatmap showing gene signature score of TAMs, MDSCs, Angiogenesis and Phagocytosis in selected macrophage subclusters as indicated. Bubble size represents the proportion of cells with enrichment gene score > 1. The colour of the circle represents the enrichment gene scores. k The ratio of interaction strength and numbers of immune cells as indicated in the immune infiltrating subtype compared to those of non-immune subtypes. Bubble size represents the ratio of interaction strength. The colour of the circle represents the ratio of interaction numbers.
To gain a deeper understanding of immune infiltration in OC, we analysed 4667 T cells from the four C2 samples and classified them into 7 subclusters (Fig. 4d). We identified two CD4+ subclusters (T02 and T03) with expression of IL7R and FOXP3 and four CD8 + T-cell subclusters (T01, T05, T06 and T07) with expression of GZMK, LAG3, MKI67 and SLP1 (Fig. 4d, Fig. S4f, g, Table S5a). We also identified an NKT cluster in the T-cell population that was marked by the expression of cytotoxic genes NKG7, GNLY and GZMB [5, 29] (Fig. 4d, Fig. S4f, g, Table S5a), consistent with the notion that NK cells transcriptionally resemble T cells. Further interrogation of the T-cell subpopulations in the immune infiltrating subtype revealed that the CD8 + GZMK T01 subcluster and the CD4 + IL7R T02 subcluster showed significantly higher percentages in the immune infiltrating subtype than in the other Proteomic subtypes (Fig. 4e, Fig. S4h). The CD8 + GZMK T01 subcluster has been characterized in several studies and is presumed to be effector memory T (Tem) cells based on its coexpression of granzymes, including GZMA, GMZH, and cytokine CCL5 [5, 30, 31]. The CD4 + IL7R T02 subcluster has been suggested to be naïve T cells with high coexpression of CCR7 [32, 33].
The effects of T cells are maintained by homoeostatic regulation of activation and exhaustion. We thus evaluated the expression of immune inhibitory checkpoint genes that determine the cytotoxic function of T cells. We found that the CD8 + LAG3 T05 subcluster, which accounted for approximately 9% of immune cells, expressed the highest levels of the PDCD1 and LAG3 genes (Fig. 4f). In addition, CTLA4 was highly expressed in the CD4 + FOXP3 T03 subcluster, which only accounted for 16% of cells in tumours. In contrast, CD8 + GZMK T01, the most abundant subcluster, expressed low levels of PDCD1 (PD1), CD274 (PDL1), PDCD1LG2 (PDL2) and CTLA4 but a moderate level of LAG3 (Fig. 4f).
Myeloid cells, which include DCs, macrophages, mast cells, and monocytes, were the second most enriched immune cells in the immune infiltrating subtype. We next employed a similar algorithm to interrogate the myeloid subpopulation and obtained 7 subclusters [5] (Fig. 4g, Fig. S4i). DCs and macrophages share overlapping molecular profiles [34, 35]. Subclusters 4 (M04) and 5 (M05) were defined as conventional DCs (cDCs) characterized by high expression of CST7, HMGA, CD1C and CLEC10A [29]. M03 was defined as monocytes based on the expression of the key markers FCN1, S100A and VCAN [5, 29]. M06 was defined as the mast cell subcluster based on the expression of the genes CSF1 and TIMP3 [29]. The remaining clusters (M01, M02 and M07) were identified as macrophages based on their high expression of the characteristic macrophage genes CD68, CD163, CCL8 and MRC1 (Fig. 4h, Fig. S4i, Table S5b). Among subclusters, the macrophage CCL8 M01 and MRC1 M02 subclusters showed higher proportions in all myeloid cells (Fig. 4i, Fig. S4j). The MRC1 M02 subcluster, which contained high expression levels of HLA-DRs (members of the MHC II family), has been suggested to have enhanced interactions with CD4 + T cells [36].
We then determined differentially expressed pathways in the myeloid compartment between C2 and C1/C3/C4. In the C2, myeloid overexpressed genes were enriched in myeloid leucocyte activation, especially T cell activation. Gene products such as C1QA, CCL5, and antigen processing via MHC class I (HLA-A, HLA-B) were mainly recognized by CD8+ T cells. In contrast, MHC class II gene products, (HLA-DMA, HLA-DMB, HLA-DOA, HLA-DPA1, HLA-DPB1, HLA-DQA2, HLA-DQB1, HLA-DRA, HLA-DRB5, HLA-DRB6) which are mainly recognized by CD4+ T cells, were enriched in C1/C3/C4 (Fig. S4l and Table S5d).
Tumour-associated macrophages (TAMs) and myeloid-derived suppressor cells (MDSCs) are distinct types of myeloid [29, 37]. Increasing evidence indicates that MDSCs are linked to tumour progression and poor prognosis [37,38,39]. We initially analysed TAM and MDSC scores across several major myeloid populations in C2 patients using scRNA-seq data (Table S5c). Our findings revealed that M03, characterized by a high MDSC score, was associated with pro-tumour property (Fig. 4j, Figure Sk). The functional status of TAMs was further evaluated based on M1/M2 polarization, which has opposing activities. While, M1 and M2 gene signatures [40] (Table S5c) can coexist within TAMs [41] which was also observed in our data (Fig. S4k). Thus, relying solely on the M1/M2 signature score may lead to inaccurate functional predictions. We then evaluated the myeloid immune response of C2 using angiogenic and phagocytic signatures (Table S5C), which were previously established to identify functional macrophages in colorectal cancer, liver cancer and pan-cancer single-cell analyses (Table S5c) [29, 41]. The phagocytosis-like macrophages (M01, M02 and M07) possessed high phagocytosis-related gene scores and were associated with anti-tumour phenotype (Fig. 4j, Fig. S4k), implying a potentially favourable antitumor immune response of myeloid cells in the C2 subtype. Immune cell-cell interaction analysis showed enhanced interactions between myeloids and T cells for patients in the immune infiltrating subtype but not in non-immune subtype patients (Fig. 4k, Fig. S4m). Notably, M01, M02 and M07 exhibited significantly increased interactions with CD8 + GZMK T01 cells in the immune infiltrating subtype compared with non-immune subtypes (Fig. 4k, Fig. S4n).
Identification of CD40 as a potential therapeutic target for the C2 immune infiltrating subtypeDespite remarkable progress in immunotherapy for various cancers [42, 43], the outcomes in EOC have been poor. The less-than-satisfactory results from clinical trials involving immune checkpoint inhibitors, such as anti-PD-L1 avelumab (NCT02718417, NCT02580058), atezolizumab (ENGOT-ov29-GCIG(ATALANTE), NRG-GY009) and anti-CTLA4 ipilimumab (NRG-GY003), prompted the exploration of alternative immunotherapeutic target in EOC [44]. To find potential immunotherapeutic targets, we conducted a comprehensive evaluation prognostic significance of immune modulator genes using the elastic-net Cox proportional hazards (CoxPH) model for each subtype [45, 46]. Our analysis identified 2 genes that exhibited a significant association with improved PFS specifically within the immune infiltrating subtype (p-value of C2 < 0.05), while showing a less pronounced impact in other subtypes (p-value > 0.1) (Fig. 5a and Table S6a). Notably, the high expression of the myeloid marker CD40 emerged as a particularly significant gene linked to enhanced survival specifically within the immune subtype.
Fig. 5: CD40 is a potential therapeutic target for the immune infiltrating subtype.
a The heatmap showing hazard ratio (HR) values of the 22 immune modulator genes calculated using elastic-net cox proportional-hazards (CoxPH) model in each subtype. Red represents higher survival risk and blue represents lower survival risk. (* p < 0.05). b CD40 protein abundance in 4 Proteomic subtypes. (Kruskal-Wallis test). c The association of CD40 protein expression with PFS in NMF C2 immune infiltrating subtype of Xiangya OC proteomics. (Log-rank test) d The association of CD40 protein expression with PFS in all patients of Xiangya OC proteomics. e The association of CD40 protein expression with PFS in immune infiltrating subtype C2 of CPTAC OC proteomic validation cohort. f Violin plot showing the expression of CD40 and CD40LG across 6 cell subpopulations in immune infiltrating and non-immune subtypes. g Violin plot showing the expression of CD40 across 7 Myeloid subpopulations in immune infiltrating and non-immune subtypes(*p < 0.05, ***p < 0.001, Wilcoxon test). h Violin plot showing the expression of CD40LG across 7 T cell subpopulations in immune infiltrating and non-immune subtypes. i Expression sig
留言 (0)