
記住我
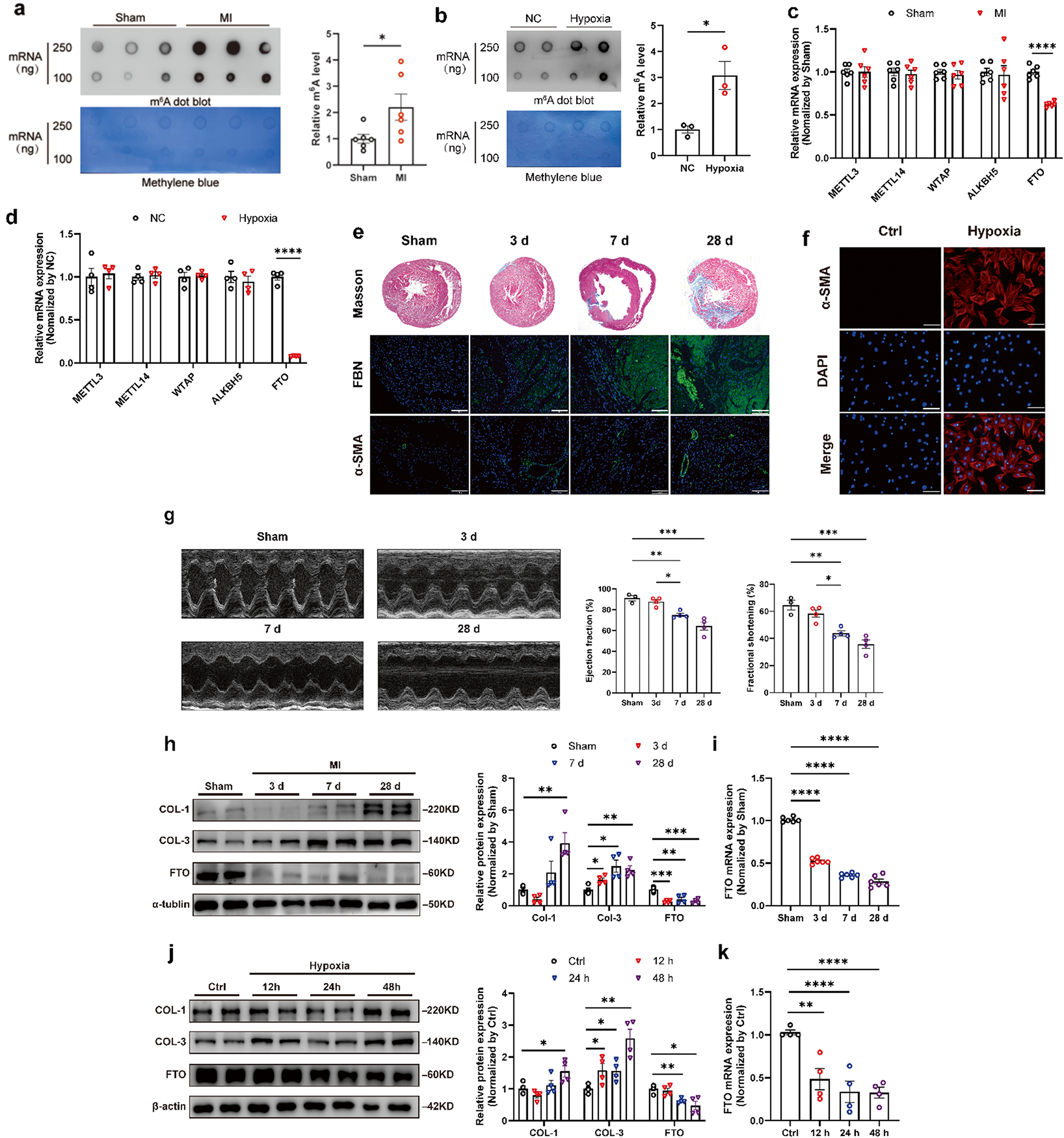



To understand the role of m6A modification in cardiac fibrosis, we assessed the expression under cardiac fibrosis in vivo and in vitro. An RNA m6A dot blot was performed to measure the levels of m6A modification. The results revealed that the m6A levels were significantly elevated in the cardiac fibrotic tissue of rats (Fig. 1a). The biological process of MI was accompanied by hypoxia. Subsequently, we established the hypoxia chamber (1% O2, 5% CO2, and balanced with N2) to explore the activation of CFs. Then we measured the m6A levels in CFs under hypoxia treatment. Consistent with the expression levels in vivo, hypoxia promoted m6A modification of CFs (Fig. 1b). The above results demonstrated that MI or hypoxia upregulated the levels of m6A modification in rats or CFs respectively. Evidence has confirmed that m6A modification levels were adjusted by methylases (METTL3, METTL14, WTAP) and demethylases (FTO, ALKBH5)(Meyer and Jaffrey 2017; Roignant and Soller 2017). To find which methylation-related enzyme caused the abnormal m6A modification, the expression levels of these m6A-associated genes were measured in MI rats and hypoxia-treated CFs (Fig. 1c, d). FTO was decreased in both MI tissues and hypoxia-induced CFs. However, the expression of METTL3, METTL14, WTAP, and ALKBH5 remained unchanged (Fig. 1c, d).
To further know the role of aberrant FTO expression in cardiac fibrosis, the cardiac fibrosis rat model was established by ligation of LAD for 3, 7, and 28 days, and the hypoxia-induced CFs model was carried out in the hypoxia chamber for 12, 24, and 48 h. It was observed that the cardiac function of MI rats significantly decreased at 7 days post-surgery and fibrotic markers, containing fibronectin (FBN) and α-smooth muscle actin (α-SMA), increased along with MI progression (Fig. 1e). Consistent with the above results in vivo, α-SMA was markedly stimulated by hypoxia for 48 h (Fig. 1f). Western blot and quantitative real-time PCR (qRT-PCR) analysis measured the negative relationship between FTO and fibrotic markers in both the fibrotic tissue of rats and hypoxia-induced CFs, including COL-1 and COL-3 (Fig. 1h, j). In the fibrotic tissue of rats, FTO expression was significantly decreased at both the mRNA and protein levels 3 days post-MI surgery (Fig. 1h, i). Similarly, FTO expression was downregulated in hypoxia-induced CFs for 24 h at both mRNA and protein levels (Fig. 1j, k).
Fig. 1
Demethylase FTO expression decreased in rat fibrotic heart tissue and hypoxia-mediated cardiac fibroblasts. RNA dot blot of m6A levels in MI rats (a) and hypoxia-induced CFs (b). The methylene blue staining served as an internal control. The mRNA expression of m6A-regulation-related genes in MI (c) and hypoxia-treated CFs (d) respectively. e Representative Masson’s trichrome and immunofluorescence images of hearts after MI. Scale bar 100 μm. f Representative histological images with immunofluorescence staining of CFs under hypoxia conditions. Scale bar 20 μm. g Representative M-mode images of MI-treated rats showing ejection fraction (EF) and fraction shortening (FS) evaluated by echocardiography. n = 3–4. h Expression of Collagen 1 (COL-1), Collagen 3 (COL-3), and FTO in a rat model of cardiac fibrosis 3, 7, and 28 days after MI. n = 4. j Expression of COL-1, COL-3, and FTO in a cellular model of fibrogenesis induced by hypoxia for 12, 24, and 48 h. n = 4. Representative FTO expression was determined by qRT-PCR in rat (i) and cellular (k) fibrosis models at different intervention times. n = 4. The data was expressed as mean ± SEM. *P < 0.05 vs. Ctrl/Sham, **P < 0.01 vs. Ctrl/Sham, ****P < 0.0001 vs. Ctrl/Sham
HIF1α bound to FTO promoter and decreased FTO expressionThe biological process of MI was accompanied by hypoxia. It was widely known that the HIF1 signal pathway was highly activated under hypoxic conditions (Ikeda et al. 2021; Sato and Takeda 2023). Thus, we detected the expression levels of the HIF signal pathway in MI rats and hypoxia-cultured CFs. Western blot and qRT-PCR results showed that HIF1α and HIF2α levels increased as FTO levels decreased in MI rat models (Fig. 2a, b). Then, hypoxia accelerated the protein and mRNA levels of HIF1α and HIF2α while inhibiting the expression of FTO (Fig. 2c, d). To identify the biological roles and molecular mechanisms of HIF1α and HIF2α, the specific inhibitors and knockdown of HIF1α and HIF2α were performed. BAY 87-2243, a HIF1α inhibitor, significantly promoted FTO mRNA levels by contrast with PT2385, a HIF2α inhibitor, remaining FTO unchanged (Fig. 2e). Then, the knockdown of HIF1α and HIF2α of CFs were established. The silencing efficacy at both mRNA and protein levels was confirmed (Fig. 2f, g). However, only HIF1α knockdown increased the FTO expression level (Fig. 2f, g). Therefore, HIF1α, rather than HIF2α, negatively regulated FTO expression.
A CUT&Tag assay was performed to elucidate the specific molecular mechanisms by which HIF1α regulated FTO expression. The results revealed that the binding sites of HIF1α were primarily located in the transcriptional start site (TSS) under normoxia and hypoxia conditions (Fig. S1a, b, c). Signal pathways correlated with DNA replication and CF activation were enriched in HIF1α-mediated CUT&Tag samples (Fig. S1d). The CUT&Tag data displayed peaks at the FTO promoter, suggesting the specific binding sites of HIF1α (Fig. 2h). Abundant evidence has confirmed that HIF-1α could bind to the promoter of target genes via hypoxia transcriptional response elements (HRE) (Liu et al. 2018; Singh et al. 2022). Then, we analyzed the sequences and found two potential HRE sites in the FTO promoter (Fig. 2i). Initially, we conducted a luciferase reporter plasmid controlled by the rat FTO promoter (-1034 bp upstream of TSS, FTO-luc). To identify the importance of these HRE sites for HIF1α-regulated FTO expression, site-directed mutagenesis was carried out to mutate the HRE motifs from ACGTG to AAGGA in the FTO-luc construct. Mutations at Site 1 and Site 2 significantly increased the HIF1α-regulated FTO expression, although Site 1 had only minor effects (Fig. 2j). The following CUT&Tag-qPCR experiments also confirmed greater enrichment at Site 2 compared to Site 1 (Fig. 2k).
In all, the above data demonstrated that HIF1α bound to the FTO promoter via HRE in CFs. MI or hypoxia treatment activated HIF1α and inhibited gene FTO transcription. Thus, HIF1α is a transcriptional inhibitor of FTO.
Fig. 2
MI-induced HIF1α accumulation negatively regulated FTO by binding to its promoter. Western blot (a) and qRT-PCR (b) results of HIF1α, HIF2α, and FTO in MI heart tissues. n = 6. The protein (c) and mRNA (d) levels of HIF1α, HIF2α, and FTO in hypoxia-treated cardiac fibroblasts (CFs). n = 4. e Expression level of FTO after treatment of HIF1α inhibitor BAY 87-2243 and HIF2α inhibitor PT2385. n = 4. Western blot (g) and qRT-PCR (f) results showing the expression of HIF1α, HIF2α, and FTO after HIF1α and HIF2α knockdown in CFs. n = 4. h Representative CUT&Tag-seq signal tracks at the promoter region of FTO. i CUT&Tag-seq predicting the potential binding sites of HIF1α and FTO promoter. j Relative luciferase activity after transfection with reporter plasmids showing the binding sites. The ratio of Firefly and Renilla luciferase values calculated the relative luciferase activity. k The CUT&Tag-qPCR results showing the degree of enrichment in predicted binding sites of the FTO promoter. n = 3. SCR, scramble sequences group. NC, negative control. The data was expressed as mean ± SEM. *P < 0.05 vs. NC/SCR, **P < 0.01 vs. NC/SCR, ***P < 0.001 vs. NC/SCR, ****P < 0.0001 vs. NC/SCR
FTO regulated collagen production and proliferation of CFs in vitroTo determine the function of FTO in CFs, we performed gain- and loss-of-function experiments by transfecting CFs of small interfering RNA targeting FTO (Si-FTO) and an overexpression plasmid of FTO (FTO). The transfection efficiencies of Si-FTO and FTO were validated by qRT-PCR (Fig. 3a). An RNA m6A dot blot assay was performed to measure the levels of m6A modification, which showed that Si-FTO increased the m6A levels while FTO decreased the m6A levels in CFs (Fig. 3b). The mRNA (Fig. 3d) and protein (Fig. 3c) levels of COL-1 and COL-3 were markedly increased after transfection of Si-FTO (100nM) under normal conditions. On the contrary, western blot and qRT-PCR analyses indicated decreased expression levels of COL-1 and COL-3 following transfection with FTO (1 µg/ml) without any treatment (Fig. 3e, f). 5-ethynyl-2’ -deoxyuridine (EdU) fluorescence staining showed that FTO knockdown significantly promoted the proliferation of CFs (Fig. 3g, k), and similar results were obtained from the CCK-8 assay (Fig. 3j). Meanwhile, compared with the scramble sequence group (SCR), CFs treated with Si-FTO markedly increased fibroblast migration indexed by the Transwell assay (Fig. 3h, l) and wound healing assay (Fig. 3i, m).
Fig. 3
FTO expression modulated collagen production and fibroblast activation in cardiac fibroblasts. a RNA dot blot of m6A levels in cardiac fibroblasts (CFs) with transfection of Si-FTO and FTO overexpression plasmids (FTO). The methylene blue staining served as a loading control. n = 5. b The relative FTO mRNA levels of CFs after transfection of Si-FTO and FTO. n = 3. The protein (c) and mRNA (d) expression of Collagen 1 (COL-1) and Collagen 3 (COL-3) after transfection of si-FTO. n = 4. The protein (e) and mRNA (f) levels of COL-1 and COL-3 after transfection of FTO. n = 4. g, k The EdU fluorescence dying assay after transfection of Si-FTO. Scale bar 50 μm. n = 4. Representative images (h) and analysis (l) of transwell assay captured at 12 h after transfection of Si-FTO. Scale bar 100 μm. n = 4. Microscope images (i) of wound healing assay captured at 0 and 24 h after transfection of Si-FTO. The relative speed of migration is shown in (m). Scale bar 100 μm. n = 4. j Cell viability after transfection of Si-FTO. n = 7. SCR, scramble sequences group. Vector, empty plasmid. The data was expressed as mean ± SEM. *P < 0.05 vs. SCR/Vector, **P < 0.01 vs. SCR/Vector, ***P < 0.001 vs. SCR/Vector, ****P < 0.0001 vs. SCR/Vector
Protective effects of FTO on MI-induced cardiac fibrosisSince the above findings indicated that FTO inhibited collagen synthesis under normal conditions, we detected the protective effects of FTO under pathological conditions. We first evaluated the protective effects of FTO on hypoxia-induced CFs. The results indicated that hypoxia stimulation increased collagen expression at both mRNA and protein levels. Consistent with the findings in normal conditions, overexpression of FTO reduced the mRNA and protein levels of collagen (Fig. 4a, b). In addition, FTO overexpression reduced hypoxia-mediated cell proliferation, as determined by the CCK-8 assay (Fig. 4c) and EdU fluorescence staining (Fig. 4d). Similarly, the migration ability of CFs was weakened as shown by the Transwell assay (Fig. 4e) and wound healing assay (Fig. 4f).
Then, we constructed an adenovirus carrying FTO to investigate the effects on MI rat models. The efficiency of adenovirus infection was confirmed (Fig. S2). After injection of FTO-overexpressing adenovirus for 7 days, the MI model was established by ligation of LAD. Interstitial fibrosis in MI rats was evaluated 4 weeks post-MI surgery. m6A levels in fibrotic tissues were significantly downregulated after FTO overexpression treatment (Fig. 4g). In addition, the lentivirus carrying shFTO was conducted for further detection. We observed that FTO overexpression therapy reduced MI-activated fibrotic markers, including collagen deposition, FBN, and α-SMA in contrast with FTO silencing accelerating fibrotic markers (Fig. 4h). Echocardiography was performed to assess the protective effects of FTO on cardiac function. Compared with the vector group, FTO overexpression markedly ameliorated the cardiac function of MI rats and collagen deposition (Fig. 4i, j). Conversely, the cardiac function and collagen production of MI rats with FTO knockdown significantly deteriorated (Fig. 4i, k).
Collectively, these results manifested that FTO inhibited collagen biosynthesis and activation of CFs. overexpressed FTO could reduce collagen deposition and ameliorate the cardiac function of post-MI rats.
Fig. 4
Effects of FTO overexpression and knockdown on hypoxia-mediated fibrosis and MI-induced cardiac fibrosis. The protein (a) and mRNA (b) levels of Collagen 1 (COL-1) and Collagen 3 (COL-3) after transfection of FTO overexpression plasmid (FTO) in cardiac fibroblasts (CFs) under hypoxia stimulation. n = 4. c Cell viability with transfection of FTO in hypoxia-induced CFs. n = 5–6. d The EdU fluorescence dying assay after transfection of FTO. Scale bar 50 μm. n = 4. e Representative images and quantitative analysis of transwell assay captured at 12 h accompanied by transfection of FTO. Scale bar 100 μm. n = 4. f Representative images of wound healing assay captured at 0 and 24 h after transfection of FTO. Scale bar 100 μm. n = 4. g RNA dot blot of m6A levels in MI rats with FTO-overexpressing virus. The methylene blue staining served as an internal control. h Representative histological images with Masson’s trichrome and immunofluorescence staining of MI heart. Scar bar 100 μm. i Representative M-mode images of MI-treated rats after overexpression of FTO and knockdown of FTO showing ejection fraction (EF) and fraction shortening (FS) evaluated by echocardiography. n = 5–6. The protein levels of COL-1 and COL-3 in fibrotic heart tissue induced by MI with FTO overexpression (j) and FTO silencing (k). Vector/shVector, the empty virus vector. The data was expressed as mean ± SEM. *P < 0.05 vs. Sham/Vector, **P < 0.01 vs. Sham/Vector, ***P < 0.001 vs. Sham/Vector. ****P < 0.0001 vs. Sham/Vector
FTO targeted EPRS in m6A-seq combined with RNA-seqRNA-seq was performed to investigate the underlying targets of FTO in cardiac fibrosis. Compared to the vector group, the results identified a total of 2929 differentially expressed genes (1547 upregulated genes, 1382 downregulated genes) in the FTO-overexpressed group. Overexpressed FTO resulted in decreased mRNA levels of collagen-related genes, including EPRS, Col1α1, Col3α1, and FBN (Fig. 5a). Furthermore, gene set enrichment analysis (GSEA) in RNA-seq indicated that collagen, collagen biosynthesis and modifying enzymes, and collagen chain trimerization were negatively enriched in the FTO-overexpressing CFs, suggesting the potential targets involved collagen synthesis (Fig. 5b). Proline is an essential amino acid for collagen production (Arif et al. 2018). HF, a febrifugine derivative, specifically inhibited collagen I synthesis and acted as a competitive inhibitor of prolyl-tRNA synthetase (Keller et al. 2012). However, the decreased expression of collagen I could be rescued by proline supplementation (Song et al. 2019). Then, the western blot was performed to elucidate the specific mechanisms of FTO-mediated fibrosis. The results indicated that HF treatment decreased hypoxia-induced and FTO-dependent collagen expression, which could be reversed with supplemental proline therapy (Fig. 5c). These findings revealed that FTO had a distinct role in proline-mediated collagen production. Moreover, Gene Ontology (GO) analysis of biological process suggested that FTO overexpression significantly suppressed cell migration and cell proliferation (Fig. S3a). Based on Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis, the MAPK, PI3K-Akt, and Wnt signaling pathways, which were closely associated with the activation of CFs, were negatively enriched in FTO-overexpressed CFs (Fig. S3b). In all, FTO suppressed the activity of CFs according to RNA-seq data.
To verify whether genes involved in collagen synthesis were differentially regulated by FTO-mediated m6A modification, m6A-seq was performed. There were 21,259 and 17,818 m6A peaks identified by m6A-seq, of which 7301 and 3860 were unique in the vector and FTO-overexpressed groups respectively (Fig. 5d). Then, the m6A consensus motif CCACC was highly enriched in the m6A peaks (Fig. 5e). Consistent with previous studies, m6A modifications were mainly enriched around the initiation and stop codon of the coding region (CDS) in Vector and FTO groups (Fig. 5g). GO analysis of biological process was also performed and indicated that FTO reduced cell cycle progression and cell migration in an m6A manner, which was consistent with RNA-seq data (Fig. 5f). Furthermore, KEGG analysis suggested that FTO overexpression remarkably reduced signaling pathways correlated with DNA replication and cell cycle (Fig. S3c). The above sequencing data confirmed that FTO regulated the activation of CFs via m6A modification. Subsequently, m6A-seq and RNA-seq were combined to find out the precise underlying targets of FTO-induced fibrosis. Genes were divided into the m6A group and the non-m6A group, depending on whether they were regulated by FTO-induced m6A modification. The results showed that the transcriptome fold change of genes in the m6A group was significantly lower and greater than that in the non-m6A group (Fig. 5h). This result suggested that FTO might downregulate the underlying gene expression levels via reducing m6A modification. Therefore, we focused on the decreased mRNA expression of genes with decreased m6A modification levels, which were highlighted in an orange circle (Fig. 5i). Among this circle, m6A-seq uncovered 3703 differential m6A peaks with decreased abundance in the FTO overexpressing group. Meanwhile, RNA-seq data identified corresponding 900 downregulated transcripts in FTO overexpression (Fig. 5i). Based on the above results and GO analysis (Collagen fibril organization), we focused on the collagen-related genes of which both m6A modification and expression levels were decreased. The above results suggested that FTO regulated collagen biosynthesis in a proline-dependent manner (Fig. 5c). Then, we screened out 15 genes involved in collagen synthesis from the overlap and discovered that EPRS may be the potential target of FTO-mediated m6A regulation due to promoting collagen translation in CFs since it was a protein that catalyzed the attachment of glutamic acid and proline to their cognate tRNAs for protein translation (Fig. 5j). qRT-PCR (Fig. 5k) and western blot (Fig. 5l, m) analysis verified that ERPS was negatively correlated with FTO.
Fig. 5
EPRS is identified as the downstream target of FTO via m6A modification. a Differentially expressed genes associated with collagen biosynthesis between Vector and FTO group in CFs (red represented up-regulation and green represented down-regulation). b Differentially gene profiles based on GSEA of FTO and Vector RNA-seq data. c Control or FTO-overexpressing CFs were treated with hypoxia, HF (100 nM), and/or proline (2 mM) for 24 h before preparation of whole-cell extracts for immunoblotting. d Number of unique and common m6A peaks in CFs of Vector and FTO groups. e Motif detected “CCACC” as the m6A consensus motif of CFs. f Down-regulated enrichment maps of biological process from m6A-seq reads in CFs. g Distribution of m6A peaks in mRNA transcripts in CFs. h Distribution of gene foldchange in RNA-seq with or without m6A modification. i The star plot revealing the distribution of genes with both differential (hyper or hypo) m6A peaks (Y-axis: fold change > ± 1, P < 0.05) and differential (up or down) expression (X-axis: fold change > ± 1, P < 0.05) in the FTO group compared with the vector group. The blue dots highlighted by an orange circle displaying down-regulated transcripts with decreased m6A levels were selected for the following investigations. j The collagen-associated genes with different m6A and mRNA expression levels shown in the heat map (red represented up-regulation and green represented down-regulation). k Relative mRNA expression of EPRS in Si-FTO- and FTO-treated CFs. n = 4. Western blot results showing the protein levels of EPRS in CFs with FTO knockdown (l) and FTO overexpression (m). n = 4. SCR, scramble sequences group. Vector, empty plasmid. The data was expressed as mean ± SEM. **P < 0.01 vs. Sham/Vector, ***P < 0.001 vs. Sham/Vector. ****P < 0.0001 vs. Sham/Vector
FTO regulated EPRS mRNA stability in m6A-dependent manners mediated by IGF2BP3The m6A modification levels of the target were modified by methylase and demethylase. Then the biological effects of m6A regulation were dependent on selective recognition of m6A sites by “readers”. IGF2BP1-3 have been reported to stabilize the mRNA levels (Huang et al. 2018; Feng et al. 2021). To identify which “reader” regulated the stability of EPRS mRNA, knockdown experiments for IGF2BP1-3, YTHDC1-2, and YTHDF1-3 were conducted. Both western blot and qRT-PCR analysis showed that the knockdown of IGF2BP3, not IGF2BP1/2, significantly reduced EPRS at both mRNA and protein levels of CFs (Fig.S5; Fig. 6a, b). Then, we further detected whether EPRS expression in FTO-knockdown CFs was affected by IGF2BP3. Silencing of IGF2BP3 remarkably inhibited the protein level of EPRS in CFs with FTO knockdown (Fig. 6c). It was realized that EPRS was the target of IGF2BP3. The following RIP-qPCR experiment confirmed that EPRS mRNA interacted with IGF2BP3 (Fig. 6d).
The m6A -seq data revealed that the m6A peak of EPRS in CDS shrank significantly with FTO overexpression (Fig. 6e). To verify the vital role of m6A modification in regulating EPRS mRNA, a luciferase reporter assay was established using either a wild-type EPRS-CDS sequence (WT) or a mutant counterpart (MUT) with altered m6A sites (Fig. 6f). While FTO levels was reduced, the relative luciferase activity in CFs with EPRS-WT plasmid significantly increased, whereas there was no significant change in CFs with EPRS-MUT plasmid (Fig. 6g). On the contrary, when FTO was overexpressed, the relative luciferase activity with EPRS-WT was downregulated (Fig. 6h). Moreover, the dual luciferase reporter assays demonstrated that knockdown of IGF2BP3 inhibited the luciferase activity of ERPS-WT plasmid through recognition of m6A sites (Fig. 6i, j). These findings suggested that IGF2BP3 regulated the expression level of EPRS through m6A modification. Next, RNA stability assays were conducted, and the results showed that IGF2BP3 deficiency promoted the degradation of EPRS mRNA (Fig. 6k). RNA degradation curves suggested that the half-life of EPRS mRNA was prolonged with FTO silencing in CFs, while IGF2BP3 knockdown could reverse the increased mRNA stability mediated by FTO silencing (Fig. 6l).
In summary, the upregulation of EPRS induced by FTO silencing can be attributed to the increased stability of EPRS mRNA, which was induced by elevated m6A modification. FTO regulated EPRS mRNA stability in m6A-dependent manners mediated by IGF2BP3.
Fig. 6
Stability of EPRS mRNA impaired by FTO-induced m6A modification via m6A reader IGF2BP3. a Relative expression of EPRS and IGF2BP3 mRNA after knockdown of IGF2BP3 in cardiac fibroblasts (CFs). n = 3. b The protein expressions of IGF2BP3, EPRS, Collagen 1 (COL-1), and Collagen 3 (COL-3) after transfection with IGF2BP3 knockdown in CFs. n = 4. c Western blot results of IGF2BP3, EPRS, FTO, COL-1, and COL-3 in CFs (control or FTO knockdown), with the absence or presence of IGF2BP3 silencing. n = 4. d m6A abundance of EPRS mRNA in cardiac fibroblasts between vector and FTO groups plotted by Integrative Genomics Viewer (IGV). e Graphical explanation for luciferase reporters showing that the wild type (full-length) or mutant (m6A motif mutated) sequence of the EPRS CDS region was inserted into the pmirGLO vector. f Relative enrichment of EPRS mRNA with IGF2BP3. The IgG group as a negative control for unspecific binding. Y-axis displaying the percentage of input for each IP sample. n = 3. Relative luciferase activity after transfection of EPRS-wild type or EPRS-mutated, with knockdown (g) or excessive (h) expression of FTO, and silencing of IGF2BP3 (i). n = 3 for g and n = 4–5 for h. j The ratio of Firefly and Renilla luciferase values calculated the relative luciferase activity in CFs (control or FTO knockdown), with the absence or presence of IGF2BP3 silencing. n = 3. k Half-life (t1/2) of EPRS mRNA determined by qRT-PCR in FTO-knockdown cells with actinomycin D and harvested at 0, 3, and 6 h. The mRNA stability normalized to the expression at 0 h. n = 3. l Half-life (t1/2) of EPRS mRNA in CFs (control or FTO knockdown), with the absence or presence of IGF2BP3 silencing. n = 3. NC, the negative control. SCR, scramble sequences group. Vector, empty plasmid. The data was expressed as mean ± SEM. *P < 0.05 vs. NC/ Vector/SCR, **P < 0.01 vs. NC/ Vector/IgG, ***P < 0.001 vs. NC/SCR/Vector, ****P < 0.0001 vs. SCR/Vector
EPRS was required for SiFTO-promoted fibrosis in CFsBased on the above data, we found that knockdown or overexpression of FTO upregulated or downregulated the expression of EPRS at mRNA and protein levels (Fig. 5k-m). To explore the role of EPRS in regulating FTO’s function, we verified the effects of EPRS on CFs. The expression of EPRS increased 24 h after hypoxia and 7 days post-MI surgery respectively (Fig. 7a). The specific siRNA for EPRS was constructed, and the transfection efficiency was validated (Fig. 7b). Knockdown of EPRS significantly reduced the mRNA and protein expression of collagen in CFs (Fig. 7b, c). Recent studies have indicated that EPRS promoted the translation of proline-rich mRNAs via enhanced translation elongation (Wu et al. 2020). Thus, HF, an inhibitor of prolyl-tRNA synthetase and collagen I synthesis, was used to explore how EPRS affected fibrosis. The results demonstrated that EPRS-dependent collagen production could be abolished by HF therapy. Furthermore, proline supplementation partially restored collagen biosynthesis, indicating that EPRS may accelerate collagen deposition through modulating proline and prolyl-tRNA synthetase activity (Fig. 7d, e). However, the protein levels of hypoxia-mediated collagen after HF and proline treatment were lower than those with hypoxia treatment alone (Fig. 7e, lanes 2, 3, 4, 10, 11, and 12). These results implied that EPRS’s promotion of collagen deposition is partially attributable to its prolyl-tRNA synthetase activity.
The TGF-β-SMAD2/3 pathway is the essential signaling pathway in cardiac fibrosis (Derynck and Zhang 2003). Western blot was used to verify whether EPRS affected fibrosis through the TGF-β-SMAD2/3 pathway. The results showed that hypoxia treatment promoted SMAD2/3 phosphorylation, which was alleviated by EPRS knockdown (Fig. 2b, lanes 1, 2, 3, 4). In addition to collagen I and III, phosphorylated SMAD2/3 was abolished by HF with or without hypoxia treatment (Fig. 2b, lanes 5, 6, 7, 8). Moreover, additional proline partially blocked the effects of HF therapy on cardiac fibroblasts (Fig. 7e, lanes 9, 10, 11, 12). These findings indicated that EPRS modulated fibrosis through the TGF-β-SMAD2/3 pathway. In addition, EPRS silencing suppressed the proliferation of CFs using EdU fluorescence staining (Fig. 7f) and CCK-8 assays (Fig. 7h). Moreover, the migration speed of CFs decreased through Transwell assays (Fig. 7g). In all, these findings indicated that EPRS played a significant role in collagen production and the activation of CFs in a proline-dependent manner.
Subsequently, to investigate the key role of EPRS as the downstream target of FTO in cardiac fibrosis, rescue experiments were conducted to detect whether EPRS silencing could reverse the effects of FTO silencing both with and without HF and proline treatment. The results showed that EPRS knockdown markedly reduced the synthesis of collagen induced by FTO knockdown at mRNA and protein levels (Fig. 7i, j). HF treatment abolished collagen production mediated by FTO silencing, regardless of EPRS knockdown. In contrast, proline supplementation restored collagen synthesis (Fig. 7k). Moreover, the high proliferation levels mediated by SiFTO were rescued by SiEPRS (Fig. 7l, m). Knockdown of EPRS also rescued the abnormal migration speed of CFs caused by SiFTO (Fig. 7n, o). These data indicated that EPRS was required for SiFTO-induced fibrosis via proline-dependent manners.
Fig. 7
EPRS is negatively related to FTO and regulates collagen deposition in a proline-dependent manner. Expression of EPRS in a cellular model of fibrogenesis induced by hypoxia for 12, 24, and 48 h and a rat model of cardiac fibrosis 3, 7, and 28 days after MI (a). n = 4. The protein (b) and mRNA (c) levels of Collagen 1 (COL-1), Collagen 3 (COL-3), and EPRS after transfection of EPRS silencing. n = 4. d, e Si-EPRS cardiac fibroblasts were treated with/without hypoxia, HF (100 nM), and/or proline (2 mM) for 24 h before preparation of whole-cell extracts for immunoblotting. f The EdU fluorescence dying assay after transfection of Si-EPRS. Scale bar 50 μm. n = 4. g Representative images of transwell assay captured at 12 h after transfection of Si-EPRS. Scale bar 100 μm. n = 4. h Cell viability after transfection of Si-EPRS. n = 4. The western blot (i) and qRT-PCR (j) results of COL-1 and COL-3 after transfection with Si-FTO or co-infection with Si-FTO and Si-EPRS. n = 4. k Cardiac fibroblasts were transfected with Si-FTO or co-infection with Si-FTO and Si-EPRS, accompanied by HF (100 nM), and/or proline (2 mM) for 24 h, followed by whole-cell extracts for immunoblotting. The CCK-8 assay (m) and EdU fluorescence dying assay (l) showing the proliferation ability after transfection of Si-FTO or co-infection with Si-FTO and SiEPRS. Scale bar 50 μm. n = 4. The representative images of transwell assay (n) at 8 h and wound healing assay (o) at 0 and 12 h indicating the migration ability of CFs induced by NC, Si-FTO, and Si-FTO with Si-EPRS. Scale bar 100 μm. n = 4. SCR, scramble sequences group. NC, the negative control. The data was expressed as mean ± SEM. *P < 0.05 vs. NC/SCR, **P < 0.01 vs. NC/SCR, ***P < 0.001 vs. NC/SCR, ****P < 0.0001 vs. NC/SCR
留言 (0)