
記住我
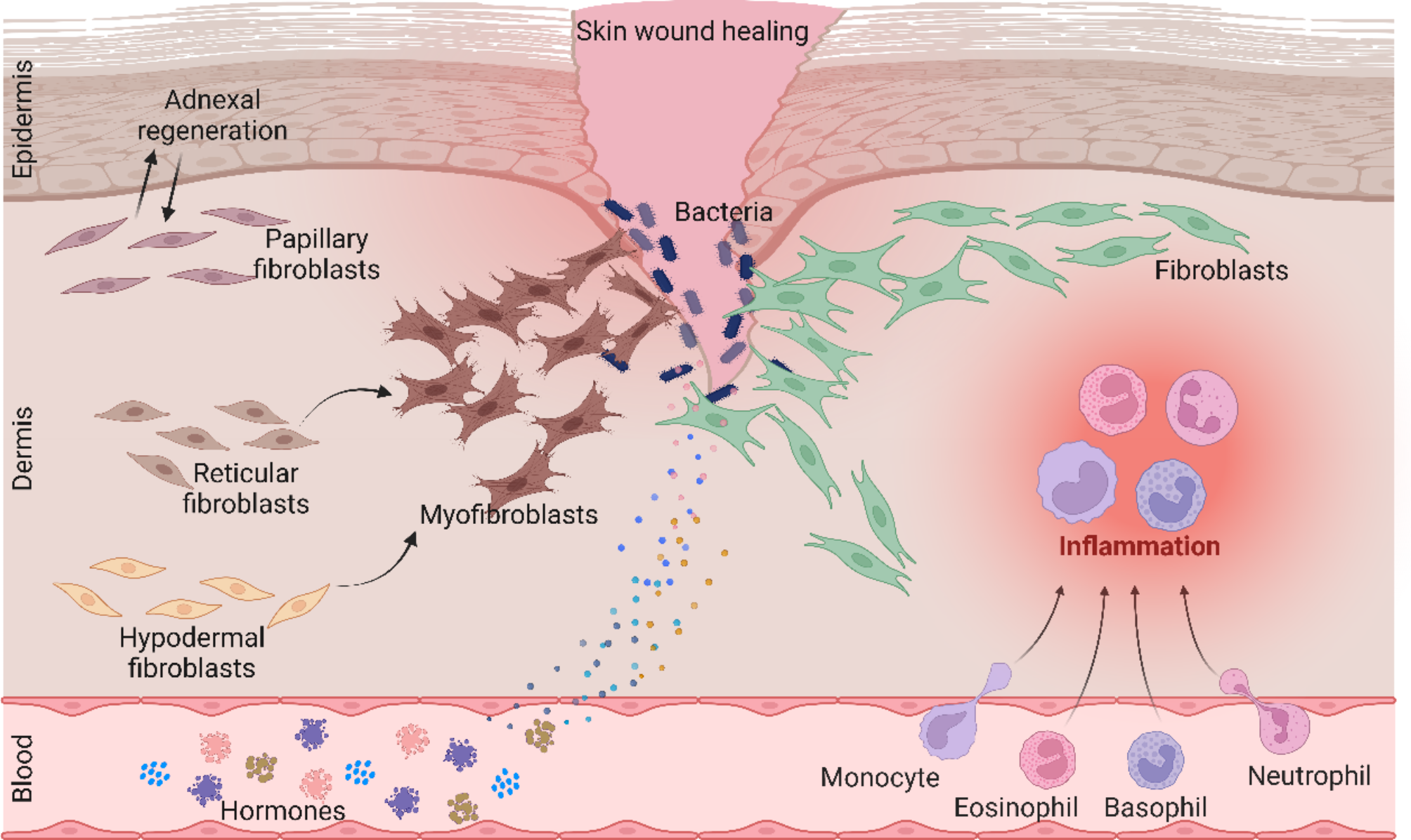



DFU is a common complication that affects the quality of life and health of millions of diabetes patients worldwide. This chronic wound is not only difficult to heal but also prone to infections and, in severe cases, may result in amputation (Bolton 2022). Despite the availability of various treatments, such as pharmacotherapy, surgical interventions, and emerging biotechnological therapies, the effectiveness of treatment varies significantly among individual patients. This variability underscores the pressing need to identify reliable biomarkers to predict treatment responses and tailor personalized medical interventions (Agidigbi et al. 2023).
With the advancement of transcriptomics technologies, we are now able to systematically analyze the gene expression patterns of DFU patients to identify key genes associated with treatment response (Wang et al. 2021). However, traditional biological statistical methods often struggle to handle the high dimensionality and complexity of transcriptome data. In this context, machine learning techniques have shown immense potential in screening genes associated with treatment response due to their exceptional data mining capabilities and pattern recognition performance. By employing machine learning methods, we can not only effectively identify potential treatment response markers from large-scale transcriptome data but also gain a deeper understanding of the molecular mechanisms underlying DFU (Wang et al. 2022b).
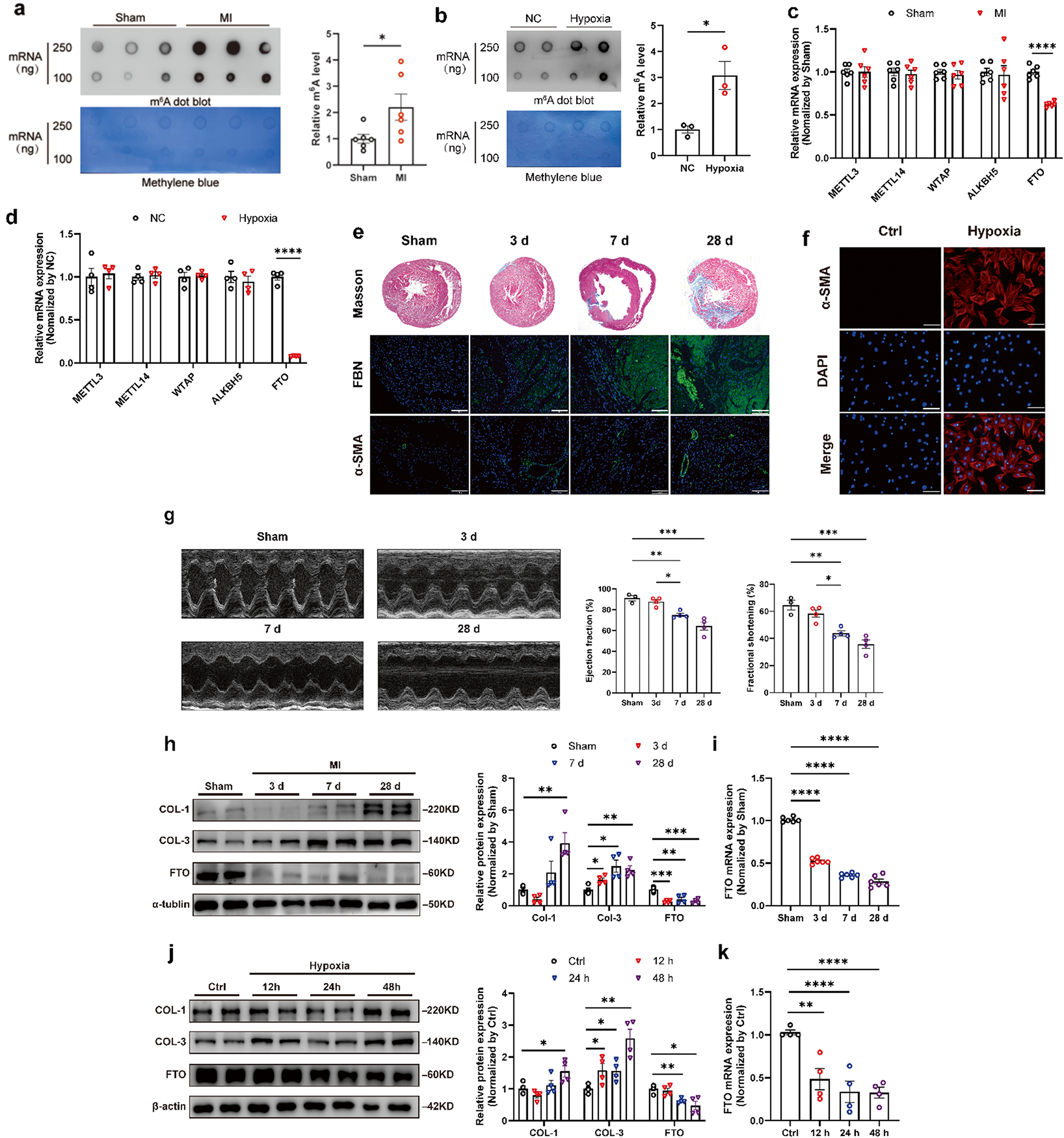
To identify key genes associated with the treatment response of DFU and provide a scientific basis for personalized treatment of DFU, we downloaded a DFU-related transcriptome dataset from the GEO database and utilized machine learning techniques to assist in the analysis of the transcriptome data. By comparing the samples from the control group at week 0 after the start of treatment with the healed samples at 8 weeks post-treatment, we conducted an in-depth exploration. The differential analysis revealed that 102 genes were significantly upregulated in the experimental group, while 548 genes were significantly downregulated (Fig. 1A). Further, GO/KEGG enrichment analysis unveiled the biological expression differences, indicating that the DEGs were mainly enriched in BP such as humoral immune response, regulation of humoral immune response, and regulation of acute inflammatory response. The CC processes showed enrichment in the extracellular region, extracellular region part, and extracellular space. Moreover, in terms of MF, significant enrichment was observed in antigen binding, glycosaminoglycan binding, and heparin binding (Fig. 1B-D). Additionally, the KEGG enrichment analysis results suggested that the DEGs were mainly enriched in pathways such as Amoebiasis, IL-17 signaling pathway, and Cellular senescence (Fig. 1E). These results indicate that the treatment response of DFU may involve multiple BP and MF, particularly pathways associated with interactions related to immune response and extracellular regions. The significant enrichment of DEGs in immune regulation, especially in humoral immune response, regulation of acute inflammatory response, and extracellular region functions, suggests that these BP may play a crucial role in the healing process of DFU. Additionally, the enrichment of these genes in MF such as antigen binding and heparin binding implies their potential involvement in pathogen recognition and immune response, which is vital for infection control and wound healing (Rong et al. 2022; Theocharidis et al. 2020). KEGG pathway analysis further reveals the importance of the IL-17 signaling pathway and cellular senescence processes, indicating that the modulation of these pathways may be key mechanisms in treatment response. The IL-17 signaling pathway plays a central role in regulating inflammation and host defense, while the involvement of cellular senescence processes may impact cell turnover and wound repair efficiency (Zhang et al. 2022). The enrichment of the Amoebiasis pathway may suggest that specific infectious pathogens or related host responses could play a role in the pathological process of DFU.
Fig. 1
Enrichment analysis reveals significant biological changes during the dfu treatment process
Note: (A) Volcano plot of DEGs in peripheral blood between pre-treatment and post-treatment healed DFU patients, with red indicating upregulated genes, blue indicating downregulated genes, and grey indicating genes with no significant difference; (B) Enriched BP in the GO enrichment analysis of DEGs; (C) Enriched CC in the GO enrichment analysis of DEGs; (D) Enriched MF in the GO enrichment analysis of DEGs; (E) Results of KEGG pathway enrichment analysis of DEGs; (F) Results of DO enrichment analysis of DEGs. The sample size for each group of patients was n = 15
Furthermore, we conducted a DO enrichment analysis to uncover the disease processes associated with the response to DFU treatment. The results revealed a strong association of numerous DEGs with certain diseases such as “bacterial infectious disease,” “leukocyte diseases,” and “hypersensitivity reaction.” These diseases demonstrated larger point sizes and lower q-values, indicating a high level of statistical significance in the enrichment (Fig. 1F). This enrichment pattern suggests that biological pathways related to these diseases may play a central role in the treatment response of DFU. Diseases like “congestive heart failure” and “atherosclerosis” were also identified in the analysis; despite involving fewer genes, their enrichment still exhibited significance, potentially reflecting a link between pathological changes in the cardiovascular system and the treatment response of DFU.
The above results indicate that DFU is closely associated with immune response, extracellular matrix interaction, and cardiovascular system changes. Furthermore, significant correlations were observed in specific disease pathways, such as bacterial infection and leukocyte diseases, providing important molecular targets and biological insights for future targeted treatment strategies.
Machine learning for selection of DFU treatment response feature genesIn order to further select characteristic genes for DFU treatment response, we utilized the Support Vector Machine-Recursive Feature Elimination (SVM-RFE) method to identify key gene expression features associated with the disease status. Based on the expression data of differentially expressed genes, we employed the rfe function for feature selection using cross-validation, incorporating SVM with radial basis kernel function and varying feature subset sizes. The analysis results were evaluated by plotting the relationship between the number of features and the RMSE of the model, indicating that all 650 differentially expressed genes met the optimal feature criteria (Fig. 2A). To further narrow down the number of feature genes, we conducted a regression analysis to fit the 650 differential genes and eliminate redundant features. The identified DFU candidate feature genes underwent Lasso regression using the “glmnet” function to model the data as a binary classification problem, with categories extracted from sample names as response variables. The model assessment involved plotting the model objects and applying cross-validation using “cv.glmnet” to determine the optimal lambda value (Fig. 2B). Ultimately, genes corresponding to non-zero coefficients extracted by the optimal lambda value were considered crucial genes related to the disease state, resulting in the final identification of 16 feature factors (Fig. 2C-D). Verification through the GeneCards online database eliminated long non-coding RNAs, pseudogenes, or other genetic elements, leading to the selection of feature genes SCUBE1 and RNF103-CHMP3. Furthermore, the prediction of their regulatory relationships and regulators was carried out using the GeneMANIA database, revealing a total of 23 regulatory factors (Fig. 2E).
Fig. 2
Machine learning algorithm for selecting feature factors in DFU treatment response
Note: (A) SVM-RFE feature curve, with the x-axis representing the number of genes and the y-axis representing the cross-validation error; (B) Distribution of LASSO coefficients for DEGs; (C) Selection of the optimal parameter (lambda) for the LASSO model; (D) Feature factors selected for DFU treatment by machine learning algorithms; (E) Network of SCUBE1, RNF103-CHMP3, and their regulatory factors, where red lines indicate physical interactions, purple indicates co-expression relationships, and yellow suggests potential interactions
Subsequently, we visualized the expression levels of SCUBE1 and RNF103-CHMP3 in the control and experimental groups, revealing significant downregulation post-treatment (Fig. S1A-B). Further investigation into their prognostic significance in DFU was conducted, plotting ROC curves which demonstrated that the AUC values of SCUBE1 and RNF103-CHMP3 were both greater than 0.8. This indicates their strong diagnostic value (Fig. S1C-D). These findings demonstrate that the machine learning algorithm successfully identified the DFU treatment response feature genes SCUBE1 and RNF103-CHMP3, which exhibit promising diagnostic potential.
To further test the diagnostic efficacy of SCUBE1 and RNF103-CHMP3, we validated their expression using the GSE80178 dataset. Our data showed that compared to non-foot ulcer samples (Non), the expression levels of SCUBE1 and RNF103-CHMP3 were elevated in diabetic foot ulcer (DFU) samples (Fig. S2A-B). Moreover, the ROC curve analysis revealed that the AUC values of SCUBE1 and RNF103-CHMP3 were both greater than 0.72, indicating a good diagnostic value (Fig. S2C-D).
The immunoregulatory role of key feature factorsIn previous analyses of BP, we observed that DFU is closely associated with immune responses, extracellular interactions, and cardiovascular changes. Moreover, they exhibit significant links in specific disease pathways, such as bacterial infections and leukocyte disorders. The immune system of diabetic patients often experiences functional impairment due to a hyperglycemic environment, affecting the functions of leukocytes, including chemotaxis, phagocytosis, and bactericidal activity, thereby rendering DFU susceptible to infections and delayed healing (Boulton et al., 2022; Ead and Armstrong 2023). Studies indicate that inflammatory cells, such as macrophages and neutrophils, play pivotal roles in the formation and healing of DFU, while the extracellular matrix provides a crucial scaffold for tissue reconstruction. Glycation under diabetic conditions and diabetes-related inflammation may disrupt normal matrix composition and extracellular signaling, thereby affecting wound healing processes (Zeng et al. 2022; Lin et al. 2022; Ead and Armstrong 2023).
The protein encoded by SCUBE1 is associated with angiogenesis and cell signaling, potentially influencing the function of vascular endothelial cells, thus playing a role in vascular changes and wound healing in diabetes (Lin et al. 2023). Proteins involved in the RNF103-CHMP3 complex may be implicated in endocytosis and the regulation of cell signaling, which could affect the release of inflammatory mediators and the immune response in wound healing (Wu and Lu 2019).
To further elucidate the impact of key regulatory factors on immune cells and activities in DFU, we conducted ssGSEA to investigate immune differences between the control group (treated 0 weeks post-DFU infection) and the experimental group (treated 8 weeks post-DFU infection, healed). The results revealed that compared to the control group, Eosinophils, Immature dendritic cells, Mast cells, and Neutrophils significantly decreased after treatment (Fig. 3A). Subsequently, we conducted an analysis of the correlation between feature genes and immune cells. The results revealed a significant negative correlation between RNF103-CHMP3 and Natural killer T cells, Type 17 T helper cells, and Type 2 T helper cells, while SCUBE1 showed a significant positive correlation with Plasmacytoid dendritic cells, Monocytes, Immature dendritic cells, and Activated dendritic cells (Fig. 3B). This outcome indicates that during the treatment of DFU, there were significant changes in the activity of specific immune cell populations, which were associated with the expression levels of key feature genes. The observed decrease in immune cells after treatment, such as Eosinophils, Immature dendritic cells, Mast cells, and Neutrophils, may reflect the positive impact of treatment in alleviating inflammation and promoting wound healing.
Fig. 3
Immunomodulatory effects of key feature factors in the healing process of DFU
Note: (A) Violin plots showing immune changes between the control group (DFU treated at 0 weeks post-infection) and the experimental group (DFU treated at 8 weeks post-infection, healed); (B) Heat map showing the correlation of SCUBE1 and RNF103-CHMP3 with immune cells. *** denotes P < 0.001 compared to the control group; ** denotes P < 0.01 compared to the control group; * denotes P < 0.05 compared to the control group
The gene RNF103-CHMP3 shows a significant negative correlation with Natural killer T cells, Th17 cells, and Th2 cells, indicating a potential inhibitory role of this gene in regulating the activity of these cell types. This may be in line with its low expression in diseased states, suggesting that during treatment and healing processes, related immune responses are weakened, contributing to a reduction in sustained inflammation. Additionally, the expression of SCUBE1 exhibits a significant positive correlation with Plasmacytoid dendritic cells, Monocytes, Immature dendritic cells, and Activated dendritic cells, suggesting that SCUBE1 may play an activating role in immune responses mediated by these cells. The downregulation of SCUBE1 after treatment may be associated with the adjustment of immune responses during treatment, aiding in reducing immune-mediated damage and promoting healing.
The results above indicate the presence of a complex interaction network between specific immune cells and key feature genes during the treatment and healing process of DFU. These genes are not only associated with expression changes during disease progression and specific immune cell activities, but their alterations may directly impact the regulation of inflammatory responses and the wound-healing process.
Expression levels of SCUBE1 and RNF103-CHMP3 in single-cell transcriptome dataPBMC samples from three subjects with cured DFU and two subjects with uncured DFU were downloaded from the GSE165816 dataset in the GEO database. The sequencing data were processed through normalization, scaling, clustering, and selection of highly variable genes. Dimensional reduction clusters are displayed in a 2D plot generated after t-SNE (PCA) clustering based on these 2000 highly variable genes (Fig. 4A). A total of 14 cell clusters were identified, and marker genes for each cell cluster are shown in Fig. 4B and Fig. S3. Further analysis revealed that SCUBE1 was primarily expressed in NK cells, with downregulated expression in NK cells of cured patients (P < 0.001) (Fig. 4C). RNF103-CHMP3 was expressed in macrophages, with reduced expression levels in macrophages of cured patients (P < 0.01) (Fig. 4D).
Fig. 4
Single-cell RNA sequencing data showing different cell types and differential expression of key genes
Note: (A) t-SNE clustering visualization shows the results of clustering single-cell RNA sequencing data using t-SNE, with different colors representing various cell types, including macrophages, NK cells, T cells, and others, totaling 14 cell types. (B) The dot plot shows the expression levels and percentages of selected genes across different cell types. The size and intensity of the color indicate the proportion of cells expressing the gene and the average expression level in the corresponding cell type. (C-D) Expression levels of SCUBE1 and RNF103-CHMP3 in single-cell transcriptome data, with the right panels showing the distribution of gene expression in different treatment groups (cured vs. uncured) in t-SNE space, where darker blue represents higher expression levels in those cells. *** indicates P < 0.001 compared to the control group; ** indicates P < 0.01 compared to the control group
留言 (0)