
記住我
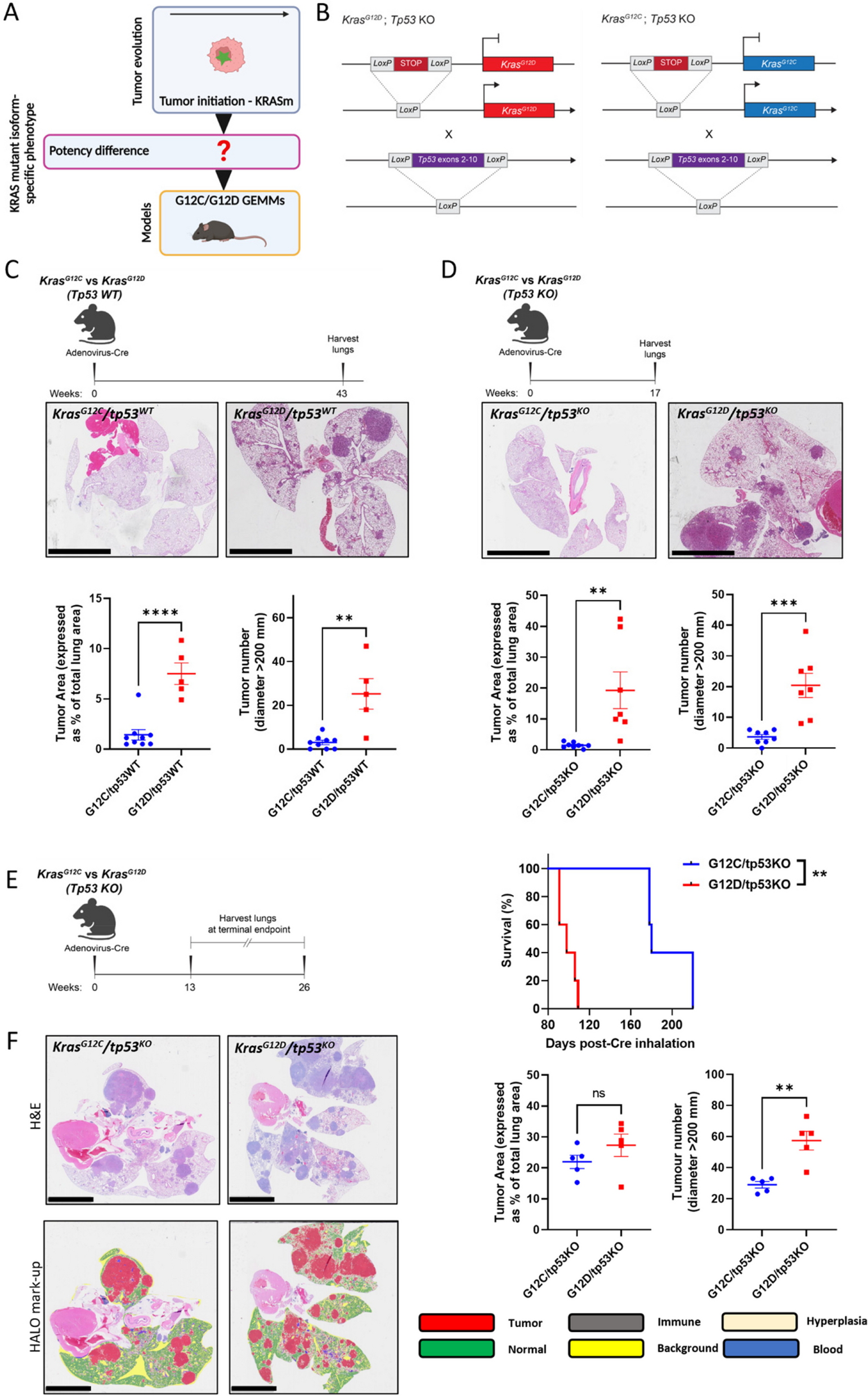

To examine and compare the oncogenic potency of KrasG12D and KrasG12C in vivo, we used a well-characterized genetically-engineered mouse model (GEMM) that harbours latent KrasG12D, whose activation with adenovirus expressing Cre recombinase (AdCre) drives the formation of lung tumors closely resembling human LUAD [21] (Fig. 1A, B). We then used CRISPR/Cas9 technology to convert the aspartic-acid-encoding codon 12 to one encoding cysteine (KrasG12C) to create a KrasG12C mouse model (Fig. 1B). Thus, we could study relative potency of KrasG12D and KrasG12C in a closely controlled in vivo setting. We further combined both Kras alleles with conditional tp53 knockout (tp53KO) to accelerate tumorigenesis and better recapitulate human NSCLC (Fig. 1B) [22].
Fig. 1
KRASG12D is more potent than KRASG12C in driving NSCLC initiation in vivo. A Schematic illustrating the use of GEMM models to study the impact of KRAS mutant isoforms on NSCLC initiation. Green star = KRAS mutation. Created with BioRender.com/b49e578. B Schematic illustrating KRAS mutant oncogenes silenced by the insertion of a STOP codon flanked by LoxP sites. AdCre administration by inhalation leads to LoxP site recombination, removing the STOP codon allowing KRAS mutant isoform expression in GEMM mice lungs. Conditional KRAS mutant mice were crossed with mice in which tp53 is also flanked by LoxP sites. AdCre induces LoxP recombination and loss of p53 protein expression. C (Above) Timeline of experiment. (Below) Representative H&E sections and HALO quantification of lung tumor area and number per mouse comparing KrasG12C/tp53WT and KrasG12D/tp53WT mice 11 months after intranasal AdCre exposure (n = 9 KrasG12C/tp53WT mice and 5 KrasG12D/tp53WT mice); scale bar = 5 mm. D (Above) Timeline of experiment. (Below) Representative H&E sections and HALO quantification of lung tumor area and number comparing KrasG12C/tp53KO and KrasG12D/tp53KO mice 4 months after intranasal AdCre exposure (n = 7–8 mice per genotype); scale bar = 5 mm. E (Left) Timeline of experiment. (Right) Survival analysis for KrasG12C/tp53KO and KrasG12D/tp53KO mice after intranasal delivery of AdCre (n = 5 mice per genotype, Log-Rank (Mantel-Cox) test). F (Top left) Representative H&E images, (Bottom left) HALO mark-up and (Right) HALO quantification of tumor area and number comparing KrasG12C/tp53KO and KrasG12D/tp53KO mice from survival study (n = 5 mice per genotype); scale bar = 5 mm. C, D and F depict mean ± s.e.m and statistical analysis carried out using unpaired Student’s t-test. ****P < 0.0001, ***P < 0.001, **P < 0.01, ns > 0.05
KrasG12C mouse models have so far been under-reported in NSCLC research, highlighting the value this tool offers to the investigation of lung cancers driven by this oncogene. We first confirmed that the KrasG12C mouse model was functional: after intranasal AdCre inhalation, lung tumors were formed which recapitulated tumors resembling lung adenocarcinoma, similarly to the KrasG12D mouse model (Supplementary Figure S1A). However, when KrasG12C were compared with KrasG12D mice matched for time after AdCre inhalation, there was a striking difference in tumorigenic properties between KrasG12D and KrasG12C models on both tp53 wild-type (tp53WT) and tp53KO backgrounds: KrasG12D-expressing mice had a dramatically increased tumor burden compared to KrasG12C (Fig. 1C, D). KrasG12D GEMMs also had increased hyperplasia compared to KrasG12C GEMMs (Supplementary Figure S1B,S1C). This difference in tumor burden was reflected by shorter median overall survival of KrasG12D/tp53KO GEMMs (98 days) compared to KrasG12C/tp53KO GEMMs (180 days) (Fig. 1E). These results were reproduced using an intratracheal method of virus inhalation with similar tumor latency observed (median survival was 211 days for KrasG12C/tp53KO mice vs 102 days for KrasG12D/tp53KO mice (Supplementary Figure S1D). However, despite a slight non-significant increase in lung tumor area for KrasG12D/tp53KO and comparable hyperplasia areas between KrasG12C/tp53KO and KrasG12D/tp53KO in mice euthanized due to disease symptoms (Fig. 1F and Supplementary Figure S1E), KrasG12D mice had approximately two-fold higher tumor number, constituted mainly by an increased number of smaller tumors (Fig. 1F and Supplementary Figure S1F). These data suggest that KrasG12D is more effective than KrasG12C in initiating NSCLC tumors.
KRASG12D co-opts the PI3K-AKT-mTOR pathway to promote tumor initiation in NSCLCWe next asked whether the increased oncopotency of KrasG12D compared to KrasG12C may be due to signaling differences immediately downstream upon tumor initiation. To investigate this, we generated an isogenic KRAS mutant initiation model using an immortalised murine lung alveolar type 2 cell line, MLE-12, that is non-tumorigenic when inoculated in mice [23], and modified it to ectopically express either flag-tagged wildtype KRAS (KRASWT), KRASG12C or KRASG12D under the control of a doxycycline-regulated promoter (Fig. 2A, B). We confirmed the correct expression of each isoform through exposure of the isogenic panel to doxycycline in the presence or absence of the KRASG12C inhibitor (G12Ci) sotorasib. Exposure to G12Ci only affected KrasG12C MLE-12 cells by binding to KRAS and switching off MAPK signaling as evidenced by reduced phosphorylated ERK. Additionally, we detected KRASG12D protein expression using a KRASG12D-specific antibody only in KrasG12D MLE-12 cells (Supplementary Figure S2A). MLE-12 cells were next cultured in ultra-low attachment (ULA) plates to induce growth as 3D spheroids, mimicking more closely the physical characteristics of cancer cells in a tumor [24]. Upon doxycycline treatment, an increase in metabolic activity and spheroid size was observed in KrasG12D- compared to KrasG12C-initiated cells, both features indicative of increased viability and proliferation respectively, consistent with our in vivo data (Fig. 2C and Supplementary Figure S2B).
Fig. 2
KRASG12D co-opts the PI3K-AKT-mTOR pathway to promote tumor initiation in NSCLC. A Schematic illustrating using GEMM models and isogenic MLE-12 cells to compare the impact of KRAS mutant isoforms on NSCLC initiation and signaling. Green star = KRAS mutation. Created with BioRender.com/b26f425. B Western blot analysis of KRAS and FLAG-tagged KRAS upon 24 h exposure of isogenic MLE-12 cells to 100 ng/mL doxycycline. PAR = parental. C (Left) MLE-12 spheroid viability upon 24 h 100 ng/mL doxycycline exposure measured by CellTiter-Glo 3D and (Right) MLE-12 spheroid area upon 96 h 100 ng/mL doxycycline exposure measured by ImageJ. Data normalised to untreated (no doxycycline) control (n = 3 at 24 h and n = 4 at 96-h). D GSEA showing that mTORC1 signaling genes are positively correlated with KRASG12D expression compared to KRASWT. E Heatmap showing DEGs belonging to mTORC1 signaling gene-set when comparing KRASG12D to KRASWT MLE-12 spheroids 24 h after 100 ng/mL doxycycline exposure (n = 3). F Western blot analysis of ERK, AKT, S6 and 4E-BP1 phosphorylation (Ser65) 24 h after exposure of isogenic MLE-12 spheroids to 100 ng/mL doxycycline. Representative of 3 independent experiments. G Flow cytometric quantification of S6 phosphorylation levels upon 24 h exposure of isogenic MLE-12 spheroids to 100 ng/mL doxycycline. Data normalised to untreated (no doxycycline) control (n = 3). H (Above) Representative immunohistochemical staining and (Below) quantification of ERK and S6 phosphorylation in early lung lesions of KrasG12C/tp53KO and KrasG12D/tp53KO mice (n = 6–9 mice per genotype); scale bar = 50 µm. I Flow cytometric quantification of surface CD44 and EpCAM in isogenic MLE-12 spheroids upon 24 h exposure to 100 ng/mL doxycycline. Data normalised to untreated (no doxycycline) control (n = 3). J Flow cytometric quantification of surface CD44 and EpCAM in isogenic MLE-12 spheroids upon 24 h exposure to 100 ng/mL doxycycline in the presence of (Left) 1 µM capivasertib (AKTi) or (Right) 1 µM ulixertinib (ERKi). Data normalised to DMSO control (n = 4). C, G, I and J depict mean ± s.e.m and statistical analysis carried out using one-way ANOVA test. (H) depicts mean ± s.e.m and statistical analysis carried out using unpaired Student’s t-test. ****P < 0.0001, ***P < 0.001, **P < 0.01, *P < 0.05, ns > 0.05. DOX = doxycycline
We next asked what could be driving the increased proliferation of KrasG12D-initiated cells. Gene expression profiling of the isogenic MLE-12 panel comparing either KrasG12D or KrasG12C to KrasWT 24 h after doxycycline exposure revealed that KrasG12D had a stronger impact on the transcriptome with 6664 differentially expressed genes (DEGs) compared to KrasWT, whereas 4566 genes were altered between KrasG12C and KrasWT (Supplementary Table S3). From these gene changes, we identified mTORC1 signaling as a significantly enriched gene-set when comparing KrasG12D to KrasWT (Fig. 2D, E), not seen when comparing KrasG12C to KrasWT, suggesting higher activation of this pathway in cells initiated with KrasG12D. Alternatively, by comparing each isogenic cell line after addition of doxycycline to its untreated counterpart, there was a strikingly higher number of differentially expressed genes belonging to the mTORC1 signaling gene-set in cells with KrasG12D expression compared to cells with KrasWT or KrasG12C (Supplementary Figure S2C). Taken together, KrasG12D expression leads to a more extensive transcriptional reprogramming, upregulating several mTORC1-associated genes compared to KrasG12C or KrasWT MLE-12 cells.
As mTORC1 is part of the PI3K-AKT-mTOR axis, a key effector pathway of RAS signaling [25], we postulated that KRASG12D co-opts the PI3K-AKT-mTOR pathway to a higher extent than KRASG12C and aimed to explore this pathway further as a potential mechanism underpinning the greater potency of KRASG12D. Firstly, we analysed if the PI3K-AKT-mTOR pathway was hyperactivated in KRASG12D-initiated cells. Indeed, higher phosphorylation of the mTORC1 activator, AKT, and the mTORC1 substrates ribosomal S6 and 4E-BP1, were evident in KrasG12D MLE-12 cells 24 h after exposure to doxycycline, consistent with our gene expression data (Fig. 2F, G). Next, using ERK phosphorylation as a measure of MAPK signaling, we observed no difference in MAPK signaling between the two mutant isoforms (Fig. 2F). By further analysing our gene expression data, we saw that DEGs related to MAPK signaling were similar between mutant isoforms (Supplementary Figure S2D). Treatment of parental MLE-12 cells with doxycycline confirmed that doxycycline does not affect either of these pathways (Supplementary Figure S2E), nor expression of exogenous KRASWT to levels matching mutant KRAS (Fig. 2F). Higher S6 phosphorylation was also noted in early KrasG12D lung lesions in vivo compared to KrasG12C lesions, whilst there was no significant difference in the level of ERK phosphorylation between the two mutant isoforms (Fig. 2H). Finally, we selected CD44 and EpCAM as two markers of NSCLC initiation [26,27,28] and showed that their expression is increased upon induction of the mutant isoforms only (Fig. 2I). Inhibition of AKT using an FDA-approved AKT inhibitor (capivasertib) [29] reduced expression of these markers to a greater extent in KrasG12D MLE-12 cells, whereas inhibition of ERK using ulixertinib reduced their expression to comparable levels in both cell lines (Fig. 2J). This suggests that the two mutant isoforms require input from the MAPK pathway for tumor initiation to the same extent, whereas the PI3K-AKT-mTOR pathway is required more during KrasG12D-driven initiation. Overall, these data confirmed our gene expression data and highlighted a possible role for the PI3K-AKT-mTOR pathway in tumor initiation by KrasG12D.
Long-term KRASG12D-exposed cells display specific PI3K-AKT-mTOR pathway dependencyHaving established the allele-specific role of KrasG12D in facilitating tumor initiation and uncovering its increased activation of the PI3K-AKT-mTOR pathway compared to KrasG12C, we next sought to explore differences between KRASG12C and KRASG12D in advanced disease to determine if growth characteristics and signaling differences are maintained during tumor evolution (Fig. 3A). As KRAS-mutant NSCLC tumors are extremely heterogenous, largely due to the diversity of their genetic alterations [9], comparing KRAS-mutant phenotypes using patient samples or NSCLC cell lines is challenging. Therefore, we began by using a panel of isogenic MEFs, initially engineered to become ‘Ras-less’ [30] and further genetically modified to express either KRASWT, KRASG12C or KRASG12D [31]. Therefore, we could compare KRAS mutant isoforms directly without the confounding effects of co-mutations seen in lung tumors. As these cells have been cultured long-term in the presence of KRAS mutant alleles, we considered that KRAS mutant isoform-dependent adaptations have developed that may reflect features of established tumors.
Fig. 3
Long-term KRASG12D-exposed cells display specific PI3K-AKT-mTOR pathway dependency. A Schematic illustrating using isogenic KRAS MEFs to determine growth rates, signaling differences and therapeutic vulnerabilities conferred by long-term expression of KRAS mutant isoforms. Green star = KRAS mutation. Created with BioRender.com/b26f425. B Spheroid area of isogenic KRAS MEFs quantified using ImageJ. Spheroid areas over time were normalised to spheroid area at day 1 (n = 3). Mean ± s.e.m. depicted. C BrdU/PI staining of isogenic KRAS MEFs at 72 h and 8 days (d8) in 3D. Proliferating cells are expressed as % BrdU-positive. Representative of three independent experiments. D Western blot analysis of ERK, AKT and S6 phosphorylation levels in isogenic KRAS MEFs at 24 h in 3D. Representative of three independent experiments. E Viability of isogenic KRAS MEFs in response to 100 nM sotorasib (G12Ci), 100 nM MRTX1133 (G12Di), 1 µM buparlisib (PI3Ki), 10 µM capivasertib (AKTi), 1 µM everolimus (mTORi) and rapamycin (mTORC1i) in 3D. Viability was measured after 72 h of drug exposure by CellTiter-Glo 3D. Viability expressed as % of DMSO control (n = 4). Mean depicted. F GSEA showing that mTORC1 signaling genes are positively correlated with KRASG12D expression and KRAS Signaling Up genes are positively correlated with KRASG12C expression. G Heatmaps showing DEGs belonging to mTORC1 Signaling and KRAS Signaling UP gene-sets comparing KrasG12C to KrasG12D MEFs (n = 3 per genotype). Statistical significance analysed using one-way ANOVA (between each time point for B or between drug treatment for E) but only significance between KrasG12C and KrasG12D MEFs presented. ***P < 0.001, **P < 0.01, *P < 0.05, ns > 0.05
First, we observed that KrasWT or KrasG12C MEFs were unable to proliferate when cultured in agarose, depriving cells of their anchorage. However, KrasG12D MEFs proliferated and formed colonies (Supplementary Figure S3A). To determine growth over time, we cultured the MEFs as spheroids in ULA plates and observed that KrasG12D MEFs were again able to proliferate. In contrast, KrasWT and KrasG12C MEFS were initially unable to proliferate, but over time KrasG12C MEFs began proliferating at the same rate as KrasG12D MEFs (Fig. 3B, C and Supplementary Figure S3B). Similar to MLE-12 cells, ERK phosphorylation levels were comparable between KrasG12C and KrasG12D MEFs after 24 h of culturing in 3D, while AKT and S6 phosphorylation were higher in KrasG12D MEFs (Fig. 3D and Supplementary Figure S3C), suggesting that KrasG12D-specific PI3K-AKT-mTOR hyperactivation persists beyond initiation. To test if the PI3K-AKT-mTOR pathway supported anchorage-independent growth of KrasG12D MEFs, we inhibited different nodes in the pathway, such as PI3K with buparlisib, AKT with capivasertib and either mTOR or mTOR complex 1 (mTORC1) with FDA-approved everolimus or rapamycin [32], respectively, and observed that KrasG12D MEFs were more sensitive than KrasWT or KrasG12C MEFs (Fig. 3E).
We next carried out gene expression profiling of KrasG12C and KrasG12D MEFs on day 8, aiming to determine differential signaling when both KrasG12C and KrasG12D MEFs were proliferating at the same rate. We observed that KrasG12C MEFs had increased gene expression associated with KRAS signaling (KRAS Signaling UP) whilst, similarly to KrasG12D MLE-12 cells, KrasG12D MEFs had increased expression of MTORC1 signaling genes (Fig. 3F, G). Collectively, these data suggest that, in cells exposed long-term to KRAS mutant isoforms, proliferation differences may become less apparent over time, however signaling differences persist as indicated by the gene expression profiles. In order to increase oncogenicity and proliferate, KRASG12C hyperactivates KRAS signaling which may be relevant to KRASG12C-driven tumor evolution. In contrast, KRASG12D relies more on PI3K-AKT-mTOR signaling, even beyond initiation, which may be therapeutically exploitable.
KRASG12C and KRASG12D NSCLC cell lines exhibit RAS effector-specific dependenciesOur above findings from the mutant KRAS MEFs led us to hypothesise that, once KRASG12C- and KRASG12D-driven tumors are established, the difference in potency between the two variants becomes less evident. Given that precision medicine and KRAS inhibitors are usually administered in the context of stage IV NSCLC, we asked whether KRAS mutant-specific dependency on the pathways described above persists in established NSCLC tumors and cell lines, conferring isoform-specific vulnerabilities (Fig. 4A). First, we carried out immunohistochemical (IHC) staining of tumors from the GEMM survival study (Fig. 1E) for markers of proliferation (Ki67), cell cycle progression (Cyclin D1) and ERK and S6 activation. Interestingly, we did not observe significant differences in the levels of staining for these proteins between KrasG12C/tp53KO and KrasG12D/tp53KO lung tumors (Fig. 4B, C) implying that there is no longer a potency (proliferation) or signaling difference between the two mutant isoforms, possibly due to acquired mutations affecting activation of these pathways [33]. In agreement, there was no significant difference in proliferation (Supplementary Figure S4) or ERK, AKT and S6 activation (Fig. 4D) between murine tumor cell lines (mTCL) derived from KrasG12C/tp53KO and KrasG12D/tp53KO GEMMs, further implying that the genotype-specific difference in potency and signaling was lost in established tumors. However, KrasG12C mTCL was more sensitive to ERK and MEK inhibition, whilst KrasG12D mTCL was more sensitive to PI3K, AKT and mTOR inhibition (Fig. 4E), despite both cell lines showing similar levels of MAPK and PI3K-AKT-mTOR pathway activation, implying that the vulnerabilities persist independently of phosphorylation levels.
Fig. 4
KRASG12C and KRASG12D NSCLC cells exhibit RAS effector-specific dependencies. A Schematic illustrating using GEMM-derived NSCLC cell lines, human NSCLC cell lines and patient data to determine growth rates, signaling differences and therapeutic vulnerabilities imposed by KRAS mutant isoforms in advanced disease. Green star = KRAS mutation. Created with BioRender.com/t58e004. B (Above) Representative immunohistochemical staining and (Below) quantification of Ki67 and Cyclin D1 expression in established lung tumors of KrasG12C/tp53KO and KrasG12D/tp53KO mice (n = 5 per genotype). Scale bar = 200 µm. C (Above) Representative immunohistochemical staining and (Below) quantification of ERK and S6 phosphorylation in established lung tumors of KrasG12C/tp53KO and KrasG12D/tp53KO mice (n = 5 per genotype). Scale bar = 200 µm. D Western blot analysis of ERK, AKT and S6 phosphorylation in KrasG12C and KrasG12D mTCLs at 48 h in 3D. Representative of three independent experiments. E Viability of KrasG12C and KrasG12D mTCLs in response to 1 µM sotorasib (G12Ci), 10 µM U0126 (MEKi), 10 µM ulixertinib (ERKi), 1 µM buparlisib (PI3Ki), 10 µM capivasertib (AKTi) and 10 nM everolimus (mTORi). Viability was measured after 48 h of drug exposure by crystal violet staining. Viability expressed as % of DMSO control (n = 3). F Western blot analysis of ERK, AKT, S6 and 4E-BP1 phosphorylation of KrasG12C and KrasG12D human NSCLC cell lines 48 h in 3D. Representative of three independent experiments. G Viability of human KRASG12C and KRASG12D NSCLC cell lines in response to 10 µM ulixertinib (ERKi) and 10 µM capivasertib (AKTi) in 3D. Viability was measured after 72 h of drug exposure by CellTiter-Glo 3D. Viability expressed as % of DMSO control (n = 3). H Cell death analyses of human (Left) KRASG12C and (Right) KRAS.G12D NSCLC cell lines in response to 10 µM ulixertinib (ERKi) and 10 µM capivasertib (AKTi) in 3D. Cell death was measured after 48 h of drug exposure by flow cytometric quantification of PI staining. Data normalised to DMSO control (n = 3). B, C, E and G depict mean ± s.e.m and statistical analysis carried out using unpaired Student’s t-test. (H) depicts mean ± s.e.m and statistical analysis carried out using one-way ANOVA ****P < 0.0001, ***P < 0.001, **P < 0.01, *P < 0.05, ns > 0.05
To determine if these findings were also relevant to human NSCLC, we first examined differences in KRAS mutant isoform-specific survival outcomes from an internationally recruited cohort of advanced KRAS-mutant NSCLC patients, the RAS-Precision Medicine (RAS-PM) database. Of 575 patients recruited, 240 were affected by cancers harboring KRASG12C, compared to 92 patients with cancers harboring KRASG12D (Supplementary Figure S5A). There were no significant differences in baseline characteristics between KRASG12C and KRASG12D mutant patients (Supplementary Table S4). There was also no significant difference in either 1st line progression-free survival (PFS) or overall survival (OS) (Supplementary Figure S5B,5C). To test whether mutant-subtype specific differences were more apparent at earlier stages of tumorigenesis in a clinical cohort, we next extracted data from cBioPortal to examine relative differences between KRASG12C and KRASG12D NSCLC (Supplementary Figure S5D) [34, 35]. We analyzed data from three cohorts: NSCLC TRACERx study (2017) [36], TCGA Firehose Legacy and Pan-Lung Cancer study (2016) [37]. In contrast to RAS-PM, this cohort was considered early stage and operable, with the intention of examining differences in RAS subtypes at point of diagnosis rather than deterioration. In line with our preclinical observations, the proportion of KRASG12D T3 and T4 stage tumors was higher compared to KRASG12C NSCLC (Supplementary Figure S5E). Taken together, these clinical results parallel our in vitro and in vivo findings, highlighting that the tumorigenic strength of KRASG12D becomes less apparent at late stages of NSCLC, whereby the oncopotency of KRASG12C appears to ‘catch up’ with KRASG12D.
Despite the loss of potency, we wondered if signaling and therapeutic differences persist in advanced human disease. We selected a panel of 6 KRASG12C and 3 KRASG12D human NSCLC cell lines. We first assessed proliferation rates among these lines and saw no significant difference in proliferation, underpinning the late-stage in vivo and patient data (Supplementary Figure S6A). Similar to established tumours in GEMMs, there was no clear differences in ERK, AKT and S6 activation between human KRASG12C and KRASG12D cell lines (Fig. 4F), again likely due to factors such as genomic heterogeneity between cell lines or Epithelial-Mesenchymal Transition influencing pathway activation [38]. Unexpectedly and contrary to the isogenic initiation model in which we observed KRASG12D-specific hyperphosphorylation of protein translation repressor 4E-BP1, we saw KRASG12D-specific downregulation of total 4E-BP1 (Fig. 4F). This ultimately has the same implication as 4E-BP1 hyperphosphorylation which is to promote protein translation [39]. We speculate that this may have developed during KRASG12D-driven tumor progression and supports the relationship between KRASG12D and mTORC1 signaling. We then mined publicly available data from the Cancer Cell Line Encyclopedia (CCLE) and compared gene expression data between KRASG12C and KRASG12D cell lines [40]. Interestingly, human KRASG12C lines were enriched for genes related to increased KRAS signaling, while human KRASG12D lines were enriched for genes related to PI3K-AKT-mTOR and specifically mTORC1 signaling (Supplementary Figure S6B), similarly to our MEF gene expression data. Subsequently, we tested the impact of MEK, ERK and AKT inhibition in these NSCLC cell lines and observed that KRASG12C lines were more sensitive to ERK or MEK inhibition, while KRASG12D lines were more sensitive to AKT inhibition (Fig. 4G and Supplementary Figure S6C). Additionally, we observed higher cell death after ERK or AKT inhibition in KRASG12C or KRASG12D cells, respectively (Fig. 4H). Altogether, these data imply that, in advanced disease, the difference in potency between these two KRAS mutant isoforms is no longer apparent in terms of proliferation and immediate signaling. However, KRASG12C and KRASG12D cells are more susceptible to MAPK and PI3K-AKT-mTOR pathway inhibition, respectively. Therefore, these mutant-subtype specific treatment vulnerabilities persist despite the loss of clear differences in oncogenic signaling and phenotype at this point of NSCLC evolution.
The clinical development of direct KRASG12C inhibitors provides our most advanced current means of inhibiting KRAS [12]. However, the clinical efficacy of KRASG12C inhibition (G12Ci) in NSCLC is hindered by intrinsic factors such as pathway re-activation and feedback/bypass pathways which often result in resistance [17, 41]. It is expected that resistance will circumvent KRASG12D inhibition (G12Di) in NSCLC, with reports of resistance mechanisms and combination strategies already emerging in colorectal cancer [42, 43]. Thus, it is vital to explore potential combination therapies to minimise resistance and maximise the potential of G12Di. Having identified the PI3K-AKT-mTOR axis as a KRASG12D-specific vulnerability, we next examined whether its inhibition combines effectively with G12Di in NSCLC. For this, we used two methods of calculating drug interaction – (i) the co-efficient of drug interaction (CDI) Eq. [44] and (ii) Bliss Synergy scoring (BSS) with SynergyFinder [45]. We also directly compared these results with the combined effect of G12Ci and AKTi to inform us whether this is a KRASG12D-specific drug combination effect. Firstly, using our isogenic MEF panel and our GEMM-derived cell lines of KRASG12C and KRASG12D-driven NSCLC, we saw that the combined effect of G12Di and AKTi was synergistic and more potent (CDI < 0.9) compared to G12Ci and AKTi in both models (Supplementary Figure S7A and B). Moreover, through conducting dose response matrices of increasing concentrations of G12Di and AKTi, we found that in our isogenic MEF panel the combined effect of G12Di + AKTi was again synergistic (BSS of 10.322) and, overall, more potent compared to that of the G12Ci + AKTi combination, where an additive effect was observed (BSS of -2.064) (Fig. 5A).
Fig. 5
KRASG12D inhibition and PI3K-AKT-mTOR inhibition synergise in KRASG12D cells. A Isogenic MEFs were treated with increasing concentrations of either sotorasib (G12Ci) or MRTX1133 (G12Di) and capivasertib (AKTi) in 3D. 72 h later, viability was measured by CellTiter-Glo 3D. Viability expressed as % of DMSO control. Drug interaction was calculated using Bliss synergy scoring (n = 3). B The same conditions and analysis as in 5A in H358 and HCC1171 (KRASG12C) and SKLU-1 and HCC461 (KRASG12D) NSCLC cell lines (n = 3). C Human NSCLCs were treated with either 1 nM (for H358, HOP62, H2030 and HCC1171), 5 nM (H1792) or 10 nM (H23) sotorasib (G12Ci) or 10 nM MRTX1133 (G12Di) (for A427, SKLU-1 and HCC461) or 10 µM capivasertib (AKTi) and a combination of both in 3D. 48 and 120 h later, viability was measured by CellTiter-Glo 3D. CI values were calculated and presented per cell line and as a mean of all cell lines per genotype (n = 3). D (Left) Timeline of experiment. (Right) Tumor weights after HCC461 cells were inoculated onto the chorioallantoic membrane of 10-day old chicken embryos and exposed to two rounds of treatment with 5 nM MRTX1133 (G12Di), 50 μM capivasertib (AKTi) or 5 μM ulixertinib (ERKi) individually or as part of combination treatments. (n = 7–16 tumors per condition). g = grams. E (Left) Timeline of experiment. (Right) Relative tumor volumes of KPARG12D tumors after inoculation onto the flanks of C57BL/6 mice which were exposed to 6 daily treatments of 10 mg/kg MRTX1133 (G12Di) or 100 mg/kg capivasertib (AKTi) individually or in combination. Tumors represented as % change in volume relative to day 1 measurements (n = 3–4 tumors per condition). D depicts mean ± s.e.m and statistical analysis carried out using a one-way ANOVA with Tukey’s post-test and a t-test for comparisons between G12Di and the combination treatment. E depicts mean ± s.e.m and statistical analysis carried out using a two-way ANOVA with Tukey’s multiple comparison’s test. ****P < 0.0001, ***P < 0.001, **P < 0.01, **P < 0.05, ns > 0.05. Note: CI = combination index. For CI analysis, points appearing above the top dotted line signify drug antagonism. Points appearing between the top and bottom dotted line signify drug additivity. Points appearing below the bottom dotted line signify drug synergism. For bliss synergy scoring (BSS), a value of less than -10 signifies drug antagonism, a value of between -10 and 10 signifies drug additivity and a value of above 10 signifies drug synergy
We next exposed two human KRASG12C NSCLC cell lines (H358 and HCC1171) and two KRASG12D NSCLC cell lines (HCC461 and SKLU-1) to increasing doses of G12Ci or G12Di and AKTi and again saw that the combination of G12Di + AKTi was either highly additive (SKLU-1 – BSS of 9.216) or synergistic (HCC461 – BSS of 10.93) (Fig. 5B). Interestingly, SKLU-1 cells are more resistant to G12Di than HCC461 cells as evidenced by the monotherapy responses but both cell lines have similar BSS, implying that co-targeting AKT is effective in both G12Di sensitive and relatively resistant settings. The combination of G12Ci + AKTi was either additive (HCC1171 – BSS of 4.214) or weakly additive (H358 – BSS of -1.49) in KRASG12C cell lines (Fig. 5B). Next, we utilised our full panel of human KRASG12C and KRASG12D cell lines, exposing them to either a combination of G12Ci + AKTi or G12Di + AKTi at two different time points to assess the immediate and longer term impact of the combinations in vitro. Using cell line-specific concentrations of either G12Ci or G12Di to elicit a comparable reduction in cell viability along with a single concentration of AKTi, the overall combined effect of G12Di and AKTi was synergistic in all three cell lines (CD1 < 0.9) (Fig. 5C) and significantly more potent (Supplementary Figure S8A) compared to G12Ci and AKTi at 48 h after drug exposure. At 120 h, synergy was maintained in KRASG12D lines implying that co-targeting KRASG12D and AKT is a durable treatment strategy. However, the combination became antagonistic (CDI > 1.1) for KRASG12C cell lines, suggesting that G12Ci and AKTi may be more effective separately (Fig. 5C and Supplementary S8B). Moreover, the reduction in viability with the combination of G12Di and AKTi was due to apoptotic cell death which was confirmed by rapid caspase-3/-7 activation (Supplementary Figure S9A and B). In order to confirm that the synergism between G12Di and AKTi was not due to off-target effects, we exposed KRASG12C MEFs to this combination in which no further benefit was seen compared to AKTi alone (Supplementary Figure S9C). To strengthen our findings, we next assessed the impact of co-targeting KRASG12D and mTORC1. To inhibit mTORC1, we used the third generation, bi-steric tool compound RMC-6272 which shares similar in vivo activity as clinical candidate RMC-5552 [46]. Despite not observing a genotype-specific sensitivity with mTORC1i as a monotherapy (Supplementary Figure S10A), with co-treatment alongside G12Di, we saw a synergistic impact in MEFs at 48 h (Supplementary Figure S10B) and in human lines at 120 h (Supplementary Figure S10C) compared to G12Ci and mTORC1i combination which was varied ranging from antagonistic to mildly synergistic. To further underpin the importance of inhibiting the PI3K-AKT-mTOR pathway to enhance KRASG12D inhibition, we co-inhibited either KRASG12C or KRASG12D and the MAPK pathway (ERKi). This resulted in a weakly additive response in KRASG12D cell lines and a stronger response in KRASG12C cell lines (Supplementary Figure S11A). We also assessed the G12Ci and ERKi combination in a relatively G12Ci resistant cell line (H2030- Supplementary Figure S11B) and saw similar BSS to the G12Ci sensitive H358 and HCC1171 cell lines, further indicating that targeting MAPK signaling is more effective in KRASG12C cell lines, but also suggesting that this combination is effective in both G12Ci sensitive and relatively resistant cell lines.
Following these in vitro experiments, we investigated whether the impact of co-targeting KRASG12D and AKT translated in vivo. For this, we inoculated the chorioallantoic membrane (CAM) of 10-day old chicken embryos with HCC461 or H358 cells and exposed the eggs to KRAS mutant inhibition and either ERKi or AKTi. For H358 tumors, neither AKTi or ERKi inhibition boosted the impact of G12Ci on reducing tumor weight. In fact, AKTi seemed to reduce the efficacy of G12Ci in this setting (Supplementary Figure S12A). Interestingly, in HCC461 tumors, G12Di had a greater impact on tumor weight reduction when combined with AKTi instead of ERKi (Fig.
留言 (0)