
記住我



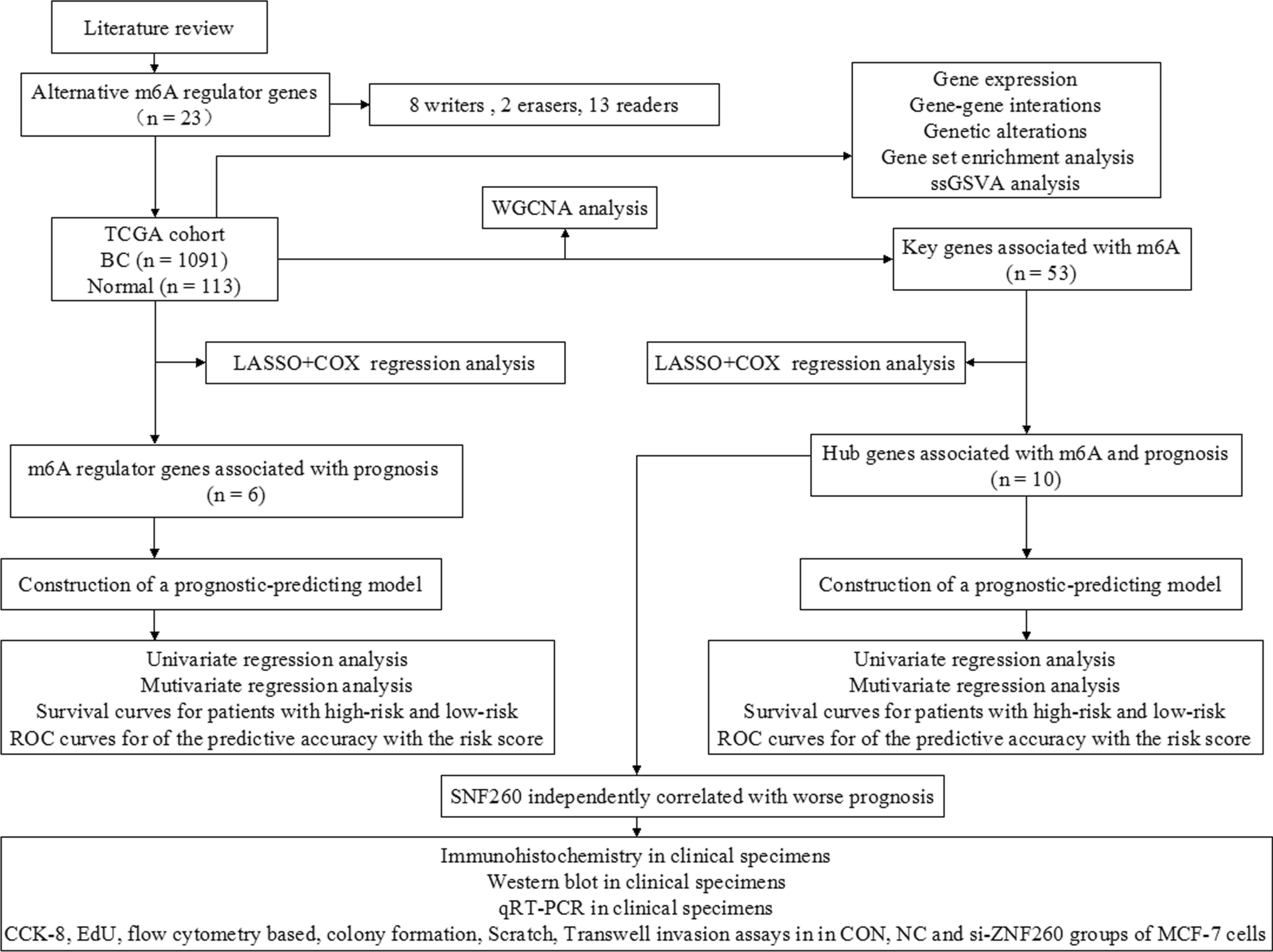
A flowchart (Fig. 1) was created to facilitate the understanding of the current study.
Fig. 1
Flowchart illustrating the study design. CON Blank control. NC Negative control
3.1 Genetic variations and expression of m6ARGs in BCTo examine the relationship between m6ARGs (writers, erasers, and readers) and breast cancer (BC), we evaluated the mRNA expression levels of 23 m6ARGs across 1,091 BC samples and 113 normal tissue samples from The Cancer Genome Atlas (TCGA). Our analysis demonstrated that 14 out of the 23 m6ARGs, such as METTL14, RBM15, KIAA1429, ZC3H13, WTAP, FTO, YTHDC1, YTHDF1, IGF2BP1, HNRNPA2B1, HNRNPC, FMR1, LRPPRC, and ELAVL1, showed differential expression between BC and normal tissues. The expression levels were visually summarized using violin plots (Fig. 2A).Somatic mutations were mainly missense variants, notably in KIAA1429, LRPPRC, FMR1, YTHDF3, and YTHDC1 (Fig. 2B). Pearson’s correlation analysis illustrated the interrelations among the 23 m6ARGs (Fig. 2C). Univariate Cox regression analysis indicated that RBM15B, ZC3H13, HNRNPC, and YTHDF3 were significantly linked to overall survival (OS) in BC patients (Fig. 2D).
Fig. 2
A Expression levels of the 23 m6A regulator genes in BC and normal tissues from TCGA. B Genetic alterations of m6A regulator genes. C Correlation matrix of the 23 m6A regulator genes. D Survival analysis of the 23 m6A regulator genes using univariate regression. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, nsP > 0.05. TCGA The Cancer Genome Atlas, m6A N6-methyladenosine, BC breast cancer
3.2 Functional and pathway enrichment analysis of m6ARGs in BCTo elucidate the functional roles and pathways involving the 23 m6ARGs, Gene Set Enrichment Analysis (GSEA) was conducted on the TCGA cohort. The results indicated that in normal tissues, m6ARGs were enriched in pathways such as adipogenesis, myogenesis, and UV response downregulation (Fig. 3A–C). Conversely, in BC tissues, m6ARGs were enriched in pathways related to cell proliferation, including E2F targets, G2/M checkpoint, MYC targets, PI3K-AKT-mTOR signaling, and glycolysis (Fig. 3D–N).This enrichment analysis provides insight into how m6ARGs contribute to the molecular landscape of BC, potentially influencing tumor growth and metastasis. Understanding these pathways could help in developing targeted therapies aimed at inhibiting these processes.
Fig. 3
Gene set enrichment analysis (GSEA) of the functions and pathways enriched in m6A regulator genes for the Normal (A–C) and BC (D–N) groups. BC breast cancer
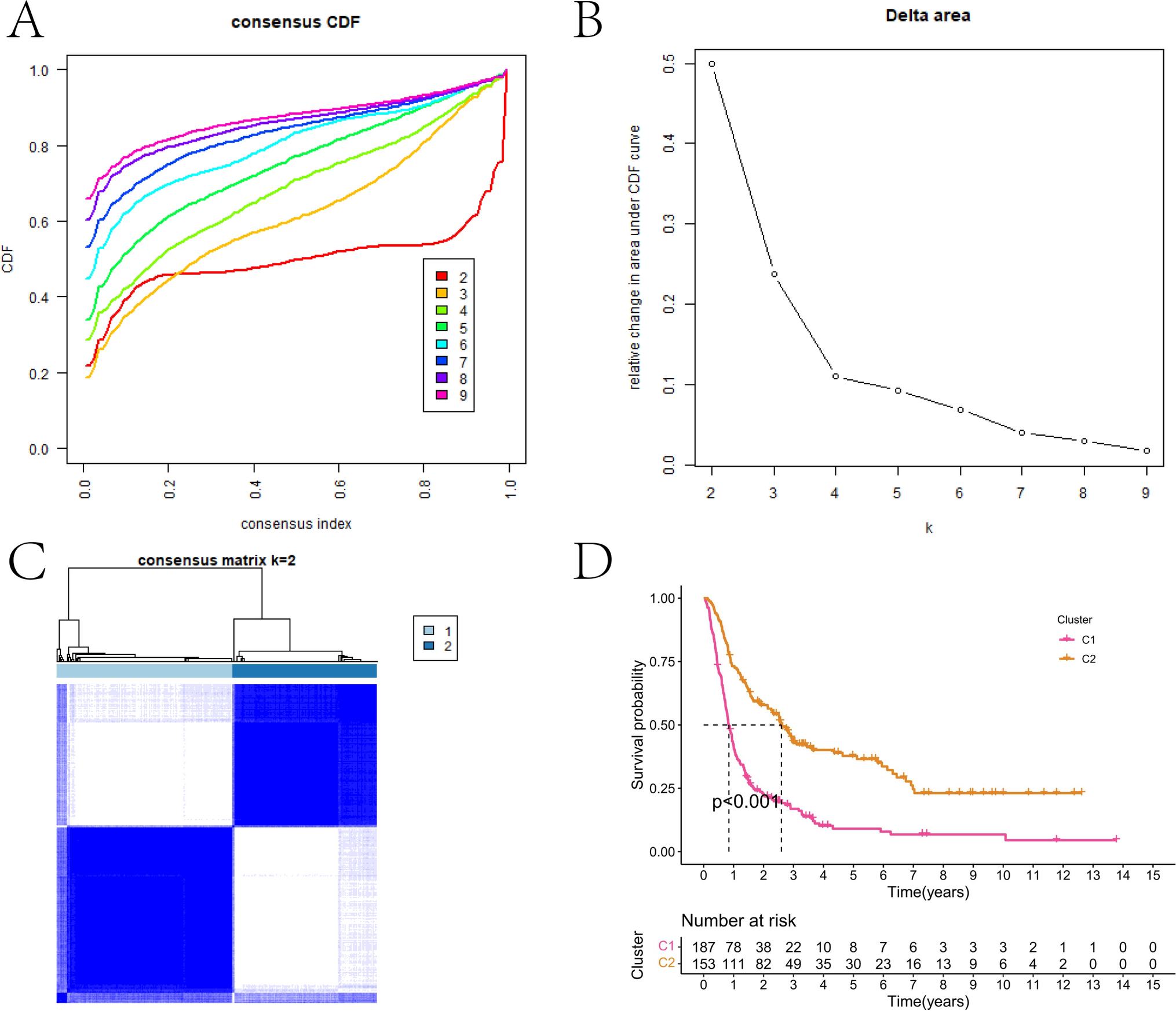
3.3 ssGSVA and BC immune cell infiltration in different m6A modification patterns in BCConsensus clustering analysis using the Consensus Cluster Plus package in R indicated that k = 2 was the optimal clustering number based on the expression profiles of the 23 m6ARGs (Fig. 4A). As a result, the 1091 BC patients were segmented into clusters 1 (n = 598) and 2 (n = 493). The expression profiles of the 23 m6ARGs were illustrated for both clusters and normal tissues (Fig. 4B). Single-sample gene set variation analysis (ssGSVA) was utilized to evaluate the biological processes within these clusters (Fig. 4C). Further analysis of immune cell infiltration revealed significant differences in T cells (CD4 memory resting, follicular helper, regulatory), macrophages (M1), dendritic cells (resting), and mast cells (resting) between the clusters (Fig. 4D). This suggests that m6A modification patterns may influence the immune landscape within BC, highlighting the potential role of m6ARGs in modulating the immune response to cancer.
Fig. 4
A Consensus clustering matrix for k = 2. B Box plots showing the expression of m6A regulator genes in normal and BC tissues across two clusters. C HALLMARK pathway enrichment scores by ssGSVA analysis across clusters. D Differences in immune cell infiltration between the two clusters. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, nsP > 0.05. BC breast cancer
3.4 Identification of prognostic m6ARGs and construction of a prognostic-predicting modelUsing LASSO + Cox regression analysis, six key m6ARGs were identified as having prognostic significance: ZC3H13, YTHDF3, YTHDF1, LRPPRC, HNRNPC, and RBM15B (Fig. 5A). These genes were incorporated into a prognostic model to compute risk scores. Patients were then divided into high- and low-risk groups based on median risk scores. Both univariate and multivariate Cox regression analyses confirmed the risk score as an independent prognostic factor (Fig. 5B and C). Patients in the high-risk category exhibited a higher mortality rate (P < 0.001, Fig. 5D).The predictive accuracy of the model was assessed via receiver operating characteristic (ROC) curves, demonstrating area under the curve (AUC) values of 0.51, 0.64, and 0.65 at 1, 3, and 5 years, respectively (Fig. 5E). This model could serve as a tool for predicting survival outcomes and guiding personalized treatment strategies.
Fig. 5
A LASSO coefficient profiles for the 23 m6A regulator genes. B Forest plot of the hazard ratio for associations between risk score, age, gender, grade, TNM stage, and overall survival in BC patients using univariate regression. C Forest plot of the hazard ratio for associations using multivariate regression. D Survival curves comparing high-risk and low-risk patients. E ROC curves assessing the predictive accuracy for 1-, 3-, and 5-year survival in the TCGA cohort. TCGA: The Cancer Genome Atlas; m6A: N6-methyladenosine; BC: Breast cancer
3.5 Identification of prognostic HBs associated with m6A and construction of a prognostic-predicting modelTo identify key modules associated with clinical traits, weighted gene co-expression network analysis (WGCNA) was applied to the TCGA dataset (Fig. 6A). A total of 796 differentially expressed genes were identified, and ten co-expression modules were distinguished. The turquoise module, containing 53 genes, was found to be strongly associated with m6A (Fig. 6B). Using LASSO + Cox regression analysis, ten hub genes (HBs) associated with m6A and prognosis were identified: USP4, PRKCDBP, GLUD1, ZNF260, PNRC2, NT5DC2, SECISBP2L, SMC3, VAPA, and STYX. These genes were integrated into a prognostic model to compute risk scores. Patients were categorized into high- and low-risk groups based on median risk scores. The high-risk group was associated with a higher mortality rate (P < 0.001, Fig. 6C). The model's predictive accuracy was assessed via ROC curves, showing AUC values of 0.70, 0.71, and 0.73 at 1, 3, and 5 years, respectively (Fig. 6D).
Fig. 6
A Clustering dendrogram of genes. B Heatmap depicting correlations between module eigengenes and the expression of the 23 m6A regulator genes. C Survival curves comparing high-risk and low-risk patients. D ROC curves for predictive accuracy of 1-, 3-, and 5-year survival in the TCGA cohort. TCGA The Cancer Genome Atlas, m6A N6-methyladenosine
This improved prognostic model demonstrates better predictive power compared to the previous model, suggesting that incorporating additional hub genes could enhance the accuracy of survival prediction in BC patients.
3.6 Relationship between ZNF260 expression and BC according to molecular typingAmong the identified genes, ZNF260 was found to be independently associated with poor prognosis (P = 0.001, HR = 1.06, 95% CI = 1.03–1.10). To validate the expression differences of ZNF260 between BC and adjacent tissues based on molecular subtypes, immunohistochemistry was performed in Fig. 7. The highest expression of ZNF260 was noted in the hormone receptor-positive/HER2-negative (HR + /HER2−) subtype BC tissues compared to adjacent tissues. Western blot and quantitative real-time PCR (qRT-PCR) analyses corroborated this observation (Fig. 8A–C).These findings suggest that ZNF260 could serve as a potential biomarker for prognosis and therapeutic targeting in specific subtypes of BC, particularly in those with HR + /HER2− status.
Fig. 7
Immunohistochemistry images showing ZNF260 expression in different molecular subtypes of BC and corresponding adjacent tissues. BC breast cancer
Fig. 8
A and B ZNF260 expression levels in different molecular subtypes of BC and corresponding adjacent tissues by western blot. C ZNF260 expression levels by qRT-PCR. qRT-PCR quantitative real-time PCR
3.7 Expression of ZNF260 in BC cell linesThe expression levels of ZNF260 were measured in three breast cancer cell lines—MDA-MB-231, MCF-7, and T47D—using western blot and qRT-PCR. Among these cell lines, MCF-7 displayed the highest expression of ZNF260, prompting its selection for subsequent in vitro experiments (Fig. 9A–C). By selecting the cell line with the highest ZNF260 expression, researchers can focus on models that best represent the conditions where ZNF260 is most active, thereby providing clearer insights into its functional role in BC.
Fig. 9
A and B ZNF260 expression levels in MDA-MB-231, MCF-7, and T47D cells by western blot. C ZNF260 expression levels by qRT-PCR. qRT-PCR quantitative real-time PCR
3.8 The effects of ZNF260 on BC cell proliferation, migration, invasion and apoptosis in vitroTo assess the impact of ZNF260 expression on breast cancer cell behaviors, si-ZNF260 was introduced into MCF-7 cells. Reduced ZNF260 expression was confirmed through western blot and qRT-PCR (Fig. 10A–C). Knockdown of ZNF260 was associated with decreased cell proliferation, colony formation, EdU-positive cells, migration, and invasion, alongside increased apoptosis (all P < 0.001) (Fig. 11A–F).
Fig. 10
A and B ZNF260 expression levels in CON, NC, and si-ZNF260 groups after transfecting si-ZNF260 and NC into MCF-7 cells by western blot. C ZNF260 expression levels by qRT-PCR. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, nsP > 0.05. qRT-PCR: Quantitative real-time PCR. CON Blank control. NC Negative control
Fig. 11
A The results of cell viability in CON, NC and si-ZNF260 groups, respectively. B The result of cell cloning in CON, NC and si-ZNF260 groups, respectively. C The result of EdU assay in CON, NC and si-ZNF260 groups, respectively. D Determination of cell migration potential by scratch assay in CON, NC and si-ZNF260 groups, respectively. E Determination of cell invasive potential by transwell assay in CON, NC and si-ZNF260 groups, respectively. F Assessment of apoptosis in CON, NC and si-ZNF260 groups, respectively. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, nsP > 0.05. CON blank control. NC negative control. EdU 5-Ethynyl-20-deoxyuridine
These findings underscore the importance of ZNF260 in breast cancer progression and highlight its potential as a therapeutic target. By inhibiting ZNF260, it may be possible to reduce tumor growth and improve patient outcomes. Further studies are warranted to investigate the mechanisms underlying these effects and to develop targeted therapies against ZNF260.
留言 (0)