2.1 Clinical samples
All clinical samples were obtained according to protocols approved by the Ethics Committee of The First Affiliated Hospital of Ningbo University. Informed consent was obtained for experiments using patient-derived tissue samples. Paraffin-embedded NPC tissues (n = 15) and normal tissues adjacent to the carcinoma (n = 15) were obtained from the Department of Pathology of The First Affiliated Hospital of Ningbo University (KY20221205). The cases were identified according to the World Health Organization histologic typing (WHOHT).
2.2 Cell culture
Human NPC cell lines (C666-1、SUNE-1、HNE-1和6-10) and NP69 human nasopharyngeal epithelial cells were obtained from the Cell Bank of Type Culture Collection of Chinese Academy of Sciences (Shanghai, China). Cells were cultured in RPMI-1640 (Invitrogen, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS, Gibco, Grande Island, USA) and 1% penicillin–streptomycin at 37 ℃ with 5% CO2.
2.3 ASRGL1 gene RNA interference lentiviral vector construction and cell infection
To determine the ASRGL1 interference gene sequences, three RNAi target sequences (GGAAGACGATCCCGAGTTCAA) were designed according to the design principles of the RNA interference sequence, DNA oligos with target sequence were synthesized, annealed, and inserted into the vector BR-V108 by Age I/EcoR I double digestion, and T4 DNA-ligase ligation. After the transformation of the ligation product into E. coli Top10 competent cells (TIANGEN, Cat. #CB104-03), the correct transformant was identified by DNA sequencing. Expression vectors and package vectors were then infected into 293 T cells, purified and the lentivirus was determined.
The NPC cell line (C666-1 and SUN-1) was infected with the obtained lentiviral particles and the transfection efficiency was assessed using ASRGL1 expression in transfected cells by quantitative reverse transcription polymerase chain reaction (RT-qPCR). The knockdown rate was evaluated, and the highest knockdown rate was selected as the target interference gene for the subsequent experiments.
2.4 Cell proliferation measurement
Cell proliferation was measured using the CCK-8 assay. C666-1 and SUN-1 cells in the logarithmic stage were harvested, digested, and resuspended, then, were seeded in 96 wells plates (2000 cells/well) and cultured. On day 1, day 2, day 3, day 4, and day 5 after transfection, 10 µl CCK-8 reagent was added to each well and cultured for 4 h. The absorbing wave at 450 nm was measured using a multiwell plate reader (Tecan infinite, M2009PR), and the number of activated cells was determined based on the OD450 value. All experiment was performed three times independently.
2.5 Cell migration measurement
Cell migration was used with the cell scratch test. The cells were seeded in 96 wells (50,000 cells/well) and incubated (37 ℃ and 5% CO2) until 90% confluence. Scratched with the sterile scratch instrument and cultured for 8 h. The wound was observed, and digital photos were taken at both the time of the scratch and after 8 h. The distance between the wound edges was measured with ImageJ software. The cell migration rate was calculated as migration rate = (distance of the initial time-distance of 8 h)/distance of the initial time. All tests were repeated three times independently.
2.6 Cell invasion measurement
Cell invasion ability was measured with Transwell assay (3422 corning). The assay was prepared, with cell plating and staining according to the manufacturer’s instructions. Briefly, Transwell inserts were placed in the well plate and incubated with serum-free medium for 2 h, serum was removed and 600 µl of 3.0% FBS was added, and transfected cells were added to the top chamber of the Transwell inserts and incubated for 24 h. Non-migrated cells were removed from the inserts and invaded cells were fixed and stained. The stained invaded cells were photo using a fluorescence microscope (Olympus, IX73), and the number of cells was obtained. The fold change was calculated by comparing the number of invaded cells in the transfected group with that in the control group. All tests were repeated three times independently.
2.7 Quantitative reverse transcription-polymerase chain reaction (RT-qPCR)
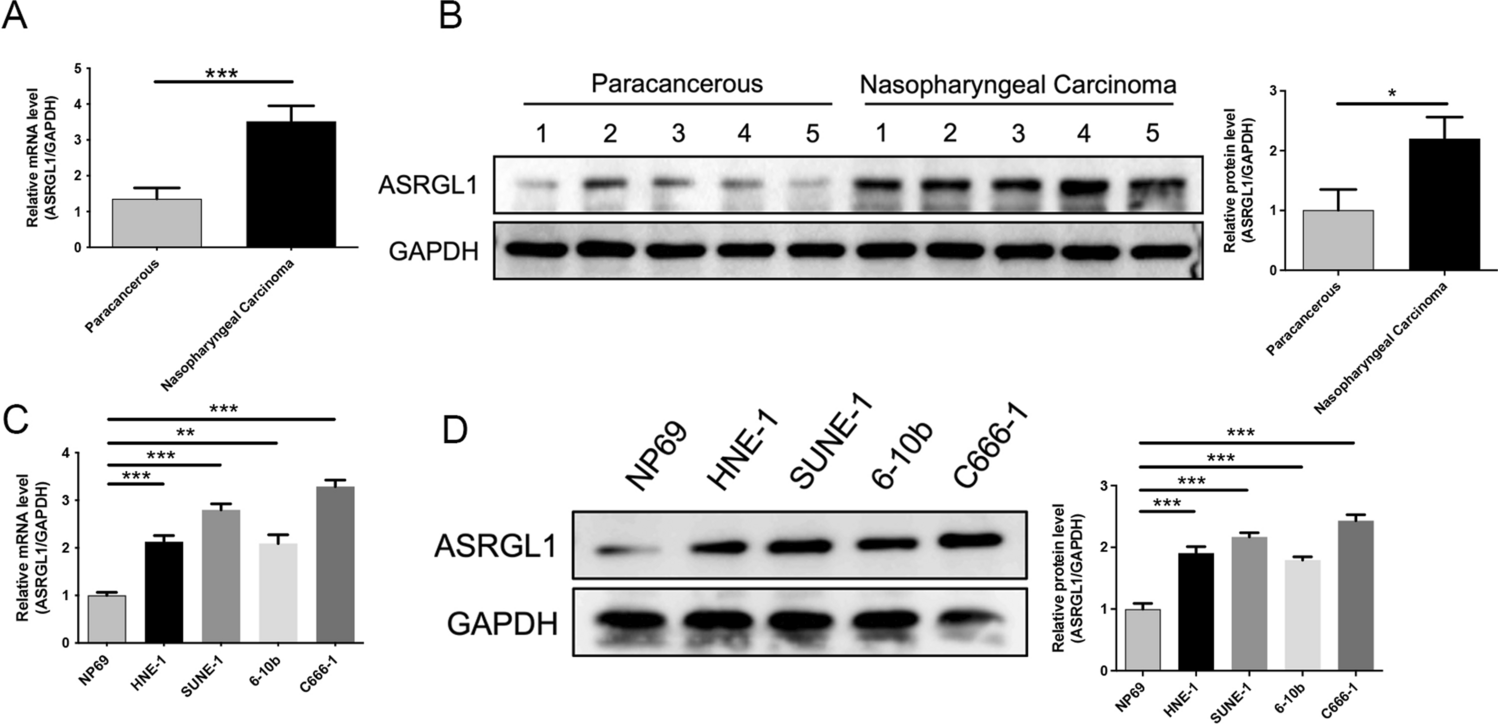
The RNA expression level of ASRGL1 in NPC cells and tissues was determined using RT-qPCR following the manufacturer’s instructions. Briefly, cells and tissues were collected, and total RNA was isolated using Trizol (Invitrogen Life Technologies, Waltham, MA, USA). Reverse transcribed cDNA using the PrimeScript RT reagent kit (Takara, Tokyo, Japan). SYBR Green Master Mix (Applied Biosystems, Foster City, CA, USA) and the ABI 7500 Real-Time PCR System were used to perform RT-qPCR analysis following the manufacturer’s instructions. 2.0 µg total RNA was synthesized to cDNA. The primers were designed as follows: GAPDH forward, 5′-TGACTTCAACAGCGACACCCA-3′ and reverse 5′- CACCCTGTTGCTGTAGCCAAA-3′; ASRGL1 forward, 5′-GGTGCTGTTGCCTTGGAC-3′ and reverse 5′-TGCCCTGTGGTTGAGACG-3′. RT-qPCR was performed, target genes were amplified and quantity, and the relative expression was analyzed with 2−∆∆Ct methods.
2.8 Flow cytometry
Cell apoptosis was detected by flow cytometry. The Annexin V-FITC kit (eBioscien, 88-8007-74) was used to segregate and quantify apoptotic cells. Apoptosis was induced with drugs. The treated cells were cultured for 5 days, digested, resuspended, centrifuged, washed with PBS and 1 × binding buffer, centrifuged, 5 µl Annexin V-APC were added to 100 μl cell suspension and incubated for 15 min, 5 μl PI and a total volume of 300 μl 1 × binding buffer was added to the tubes. The samples were analyzed by fluorescence-activated cell sorting immediately. Double-positive propidium iodide and annexin-V staining was assigned as late-stage apoptotic cells, and double-negative staining was assigned as viable cells. Early apoptotic cells were stained with annexin-V staining, and necrotic cells were stained with PI only. Total apoptosis includes early and late-stage apoptosis. All experiment was performed three times independently.
2.9 Cell cycle analysis
The cell cycle was completed by flow cytometry. Quantitation of DNA content was used to estimate the percentage of cell population in the different phases of the cell cycle. Cells infected with ASRGL1 shRNA lentivirus or control were cultured for 5 days up to 80% confluence, then digested, centrifuged, washed with PBS, fixed in cold 70% ethanol at 4 ℃ 1 h, fixed cells were stained with propidium iodide (PI) in the presence of RNase A. The cell cycle distribution was analyzed using Millipore Guava easy Cyte HT flow cytometer, and the percentage of cells in each phase of the cell cycle was quantitated using an algorithm.
2.10 Western blot protein measurement
The protein expression was detected by Western blot. NPC cells or tissues were washed and lysed with 180 μl fresh buffer made up of Western and IP Lyse, and PMSF on ice for 15 min. The lysates were then centrifuged, and the supernatants were collected to quantify protein concentration using the BCA assay. Equal amounts of proteins (20 µg) were loaded and separated by SDS-PAGE electrophoresis, and then transferred to polyvinylidene fluoride membranes (PVDF). The membranes were blocked with 5% nonfat dried milk in TBS-T with 0.1% Tween-20 for 60 min at room temperature, and then were incubated overnight at 4 ℃ with primary antibodies, including rabbit anti-ASRGL1 (ab150832, Abcam), anti-Cleaved caspase3 (ab214430, Abcam), anti-Cleaved caspase9 (9509, CST), anti-ACSL4 (ab155282, Abcam), anti-FTH1 (ab75972, Abcam), anti-GPX4 (ab125066, Abcam) and anti-GAPDH (AP0063, Bioworld). The next day, the membranes were incubated with an anti-rabbit antibody conjugated to horseradish peroxidase (HRP) (1:3,000) for 1 h. Immunoreactivity was detected using an enhanced chemiluminescence (ECL) detection system. The intensity of protein bands was quantified using Image J software (Broken Symmetry Software, USA), and protein expression levels were normalized using GAPDH as standard protein expression. All tests were repeated three times independently.
2.11 Intracellular ROS Measurement
ROS accumulation within cells was quantified. In summary, cell groups were incubated with 10 μmol/L DCFH-DA at 37 ℃ for half an hour. Using a Zeiss Fluorescence Microscope (Jena, Germany), DCF fluorescence was observed at 488 nm excitation and 525 nm emission.
2.12 ATP level
The levels of ATP were measured using the ATP Assay Kit (Beyotime, Shanghai, China). Cells (5 × 105) were introduced to the kit working solution in compliance with the manufacturer's instructions. Next, a spectrophotometer (Multiscan MK3, Thermo Fisher Scientific, Waltham, MA, USA) was used to measure the samples' absorbance.
2.13 Statistical analysis
Data were statistically analyzed using GraphPad Prism 9.0. All values are presented as mean ± standard deviation (SD). The discrepancies between the ASRGL1 knockdown group and the control group were compared with the unpaired t-test. When comparing more than two groups, two-way ANOVA was used, followed by Tukey’s multiple comparison post hoc test., p < 0.05 was considered a statistically significant difference.




留言 (0)