
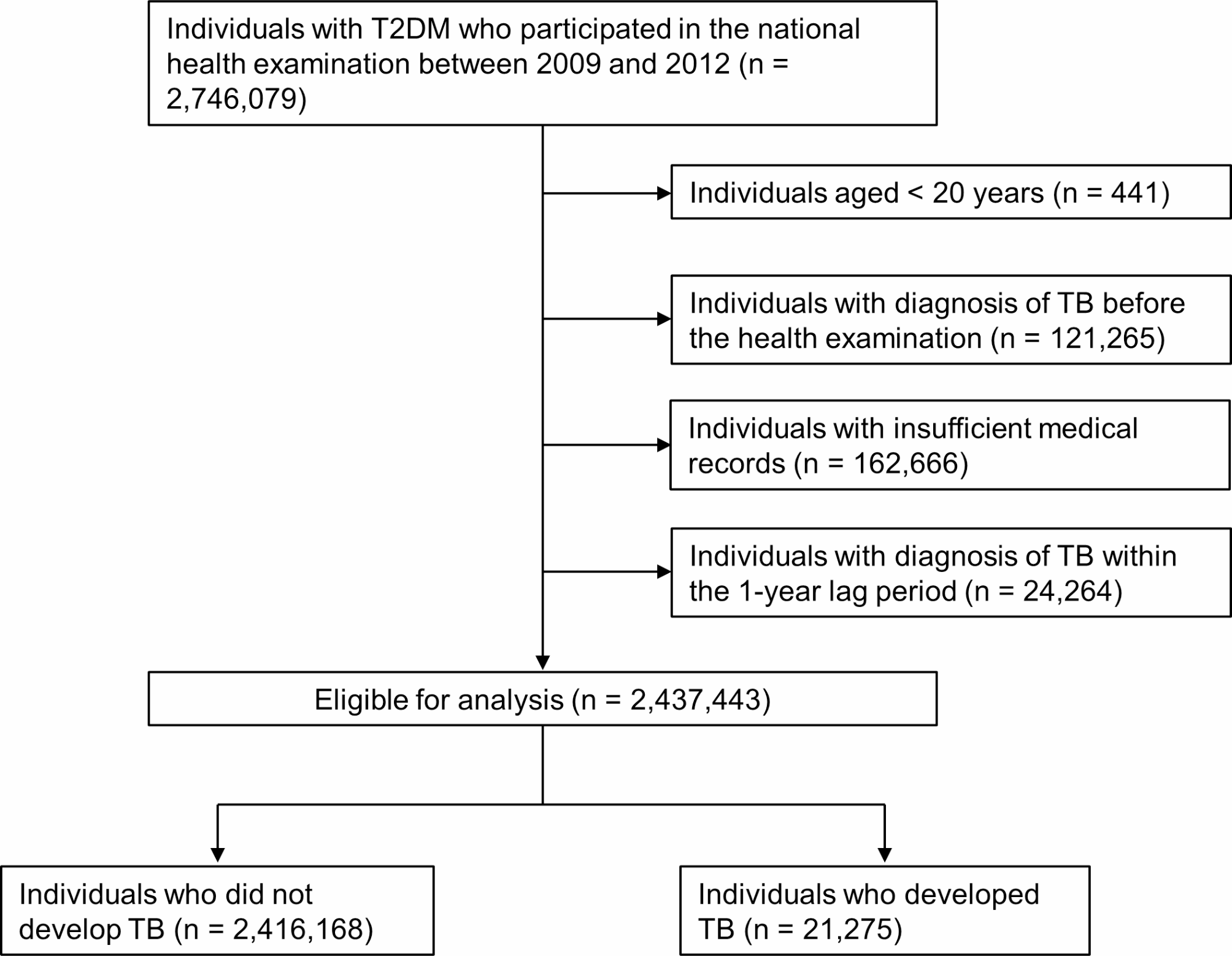
記住我
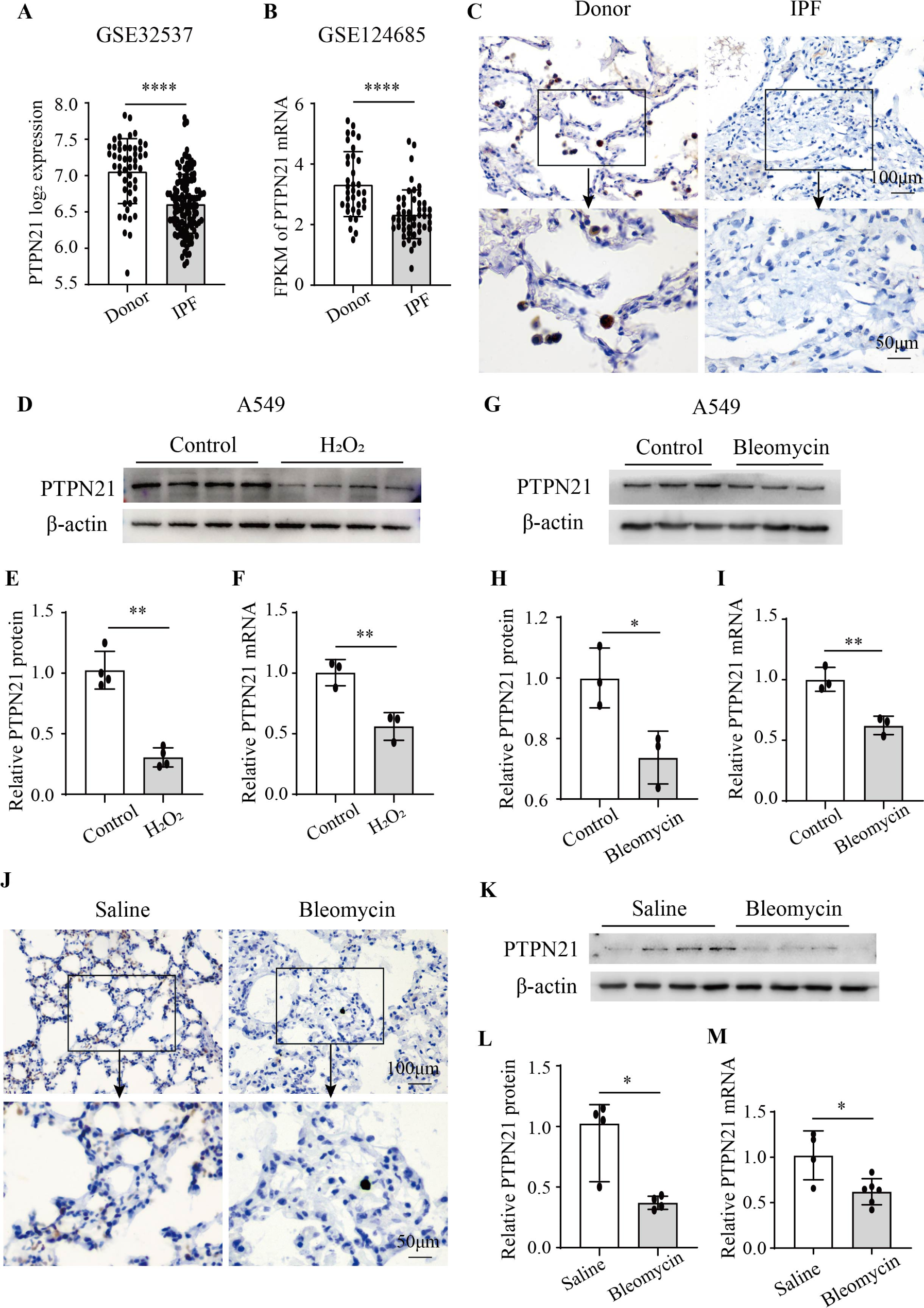
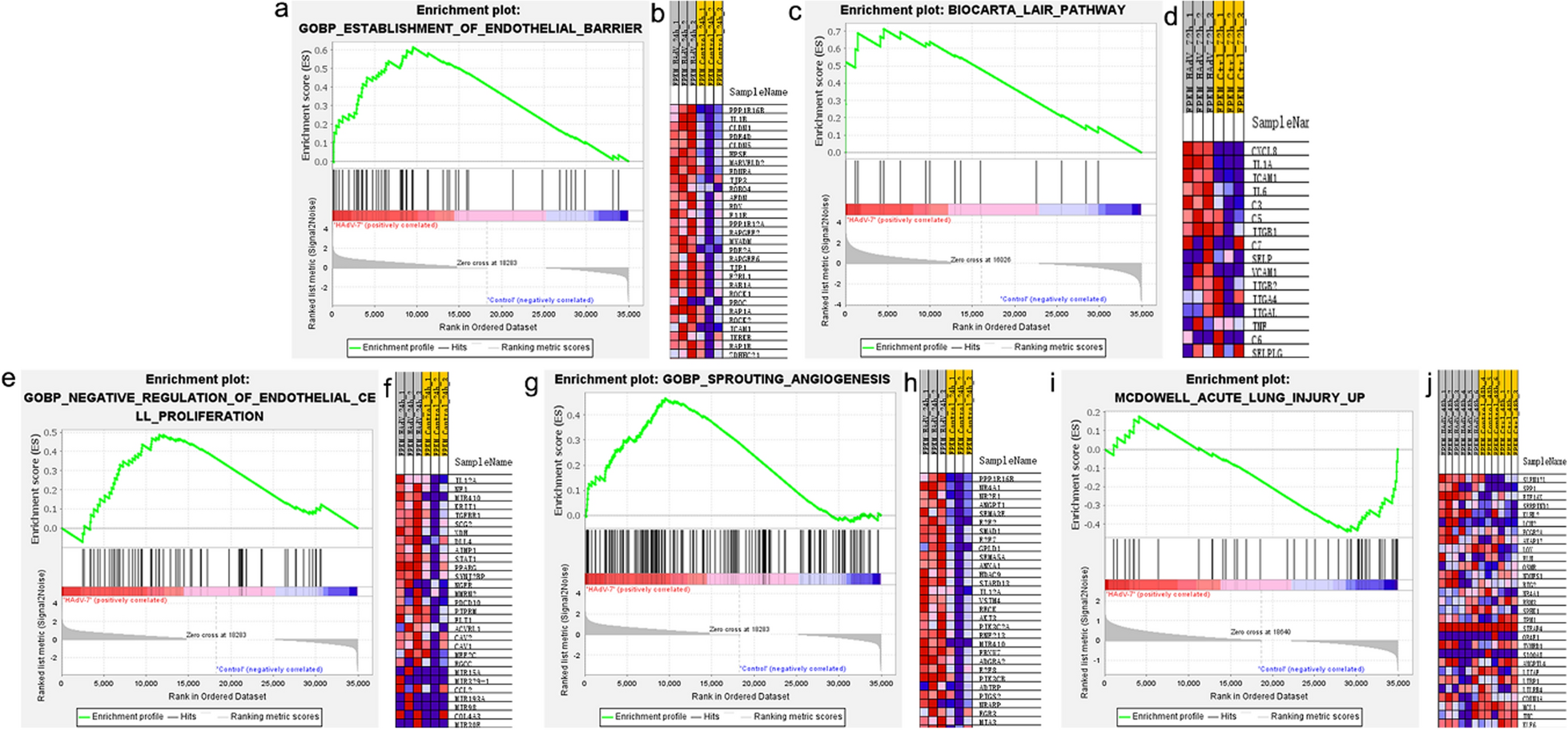
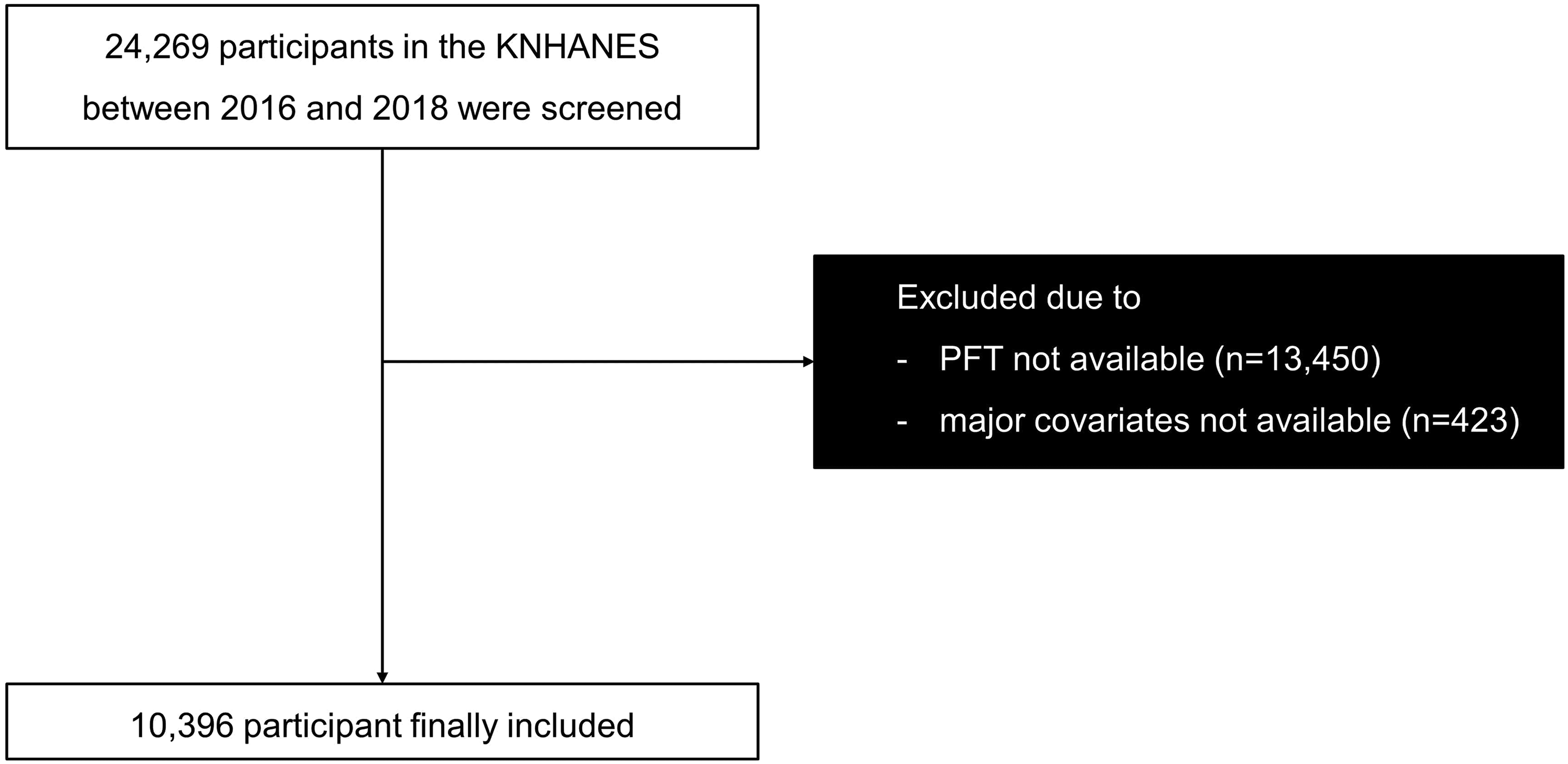
To investigate the alternations of cholesterol metabolism after hyperoxia exposure, a total of 65 infants born at < 32 weeks gestation who needed respiratory support were enrolled in this study, and clinical features were summarized in Table 1. The serum TC levels of the preterm infants who developed BPD (n = 40) at 1 week after birth were significantly higher than that of the preterm infants who did not develop BPD (n = 25), while there was no significant difference in the serum TC levels of the preterm infants at 2 weeks after birth (Fig. 1A). Meanwhile, the median inhaled FiO2 was significantly higher and the proportion of mean FiO2 > 21% was higher in the BPD group, suggesting that hyperoxia exposure may cause cholesterol metabolism disorder in preterm infants, which may be related to the development of BPD.
Table 1 The clinical features of the BPD and non-BPD infantsFig. 1
Hyperoxia exposure increased serum total cholesterol level and disrupted lung cholesterol homeostasis. (A) serum total cholesterol (TC) levels and Fraction of inspiration O2 concentration (FiO2) of the preterm infants who developed bronchopulmonary dysplasia (BPD) (n = 40) or not (n = 25). (B) Representative photographs of HE-stained lung sections from rats exposed to room air (RA group) or hyperoxia (BPD group). Scale bars, 100 μm (100X) and 50 μm (200X), n = 5/group. (C) Representative pictures of the rats and lung tissues, and the quantification of the HE-stained lung sections of the rats, shown as mean linear intercept (MLI) and radial alveolar counts (RAC), n = 5/group. (D) Weight, serum TC, lung TC content, and bronchoalveolar lavage fluid (BALF) APOA of the BPD rats and RA group. n = 8–10/group. (E) Relative mRNA expression of Abca1, Abcg1, Ldlr, Hmgcr, normalized to GAPDH. n = 6/group. The box represents mean ± SD/mean ± SEM and the whiskers represent maximum and minimum values. The experiments were repeated at least 3 times. * P < 0.05; ** P < 0.01; *** P < 0.001; **** P < 0.0001
We subsequently established a BPD rat model through hyperoxia exposure to delve deeper into cholesterol metabolism. Using HE staining, we evaluated pathological alterations in lung tissue, quantified by the mean linear intercept (MLI) and radial alveolar counts (RAC). As previously reported [24], the BPD rat model induced by hyperoxia exposure exhibited features of alveolar enlargement, evidenced by higher MLI values and lower RAC (Fig. 1B and C). Furthermore, in the BPD rat group, the lung volume was diminished (Fig. 1C). When compared to the control group under the normoxia environment, the BPD group exhibited a significant decrease in body weight, along with elevated levels of plasma TC and pulmonary TC content (Fig. 1D). Additionally, the levels of serum TC were found to correlate with oxygen concentration administered (Supplementary Fig. 1). ELISA analysis revealed that the levels of bronchoalveolar lavage fluid (BALF) APOA, which is associated with RCT, were notably reduced in the BPD group (Fig. 1D). Next, to further investigate the disrupted lung cholesterol homeostasis caused by hyperoxia, lung RNA was isolated from both groups to target genes related to cholesterol influx, efflux, and synthesis. The results indicated a significant decrease in Abca1 and Abcg1 mRNA expression, involved in RCT, in the BPD group compared to the RA group (Fig. 1E). A trend toward decline with no significant difference in Hmgcr mRNA expression indicating cholesterol synthesis was observed while there was no significant difference in mRNA level of Ldlr responsible for cholesterol intake in the BPD rat lung tissue (Fig. 1E). These above data suggested that hyperoxia exposure may also lead to lung cholesterol overload, which may be linked to circulating cholesterol metabolism disorder, contributing to BPD development.
Hyperoxia exposure led to intracellular cholesterol accumulation and oxidative stressTo further evaluate the effect of hyperoxia exposure and exogenous cholesterol overload on lung tissue, we stimulated murine lung epithelial cells (MLE12) with cholesterol at different concentrations under a hyperoxia environment in vitro. Our findings revealed an increase in intracellular cholesterol content due to external cholesterol stimulation. Additionally, hyperoxia exposure also up-regulated intracellular cholesterol accumulation (Fig. 2A), which indicated that both a cholesterol-overload environment and hyperoxia exposure have a synergistic impact on cellular cholesterol accumulation. Cellular cholesterol homeostasis is associated with mitochondrial oxidative injury [25], we next analyzed ROS production in MLE12 cells. Although with no statistical difference, we also noted a combination effect of ROS production under exogenous cholesterol and hyperoxia stimulation, but there were no differences in ROS production between different cholesterol concentrations under the normoxia environment (Fig. 2B). It revealed that hyperoxia exposure upregulated the accumulation of intracellular cholesterol, which may be associated with cellular oxidative stress, while exogenous cholesterol exacerbated this condition.
Fig. 2
Hyperoxia exposure leads to intracellular cholesterol accumulation and increased ROS production. (A) Representative pictures of MLE12 cells exposed to normoxia (NOX) or hyperoxia (HYX) for 24 h with or without 100 ¼M cholesterol stained by Filipin III and the quantification of the mean fluorescence intensity of Filipin III staining. Scale bars, 50 μm, n = 5/group. (B) Reactive oxygen species (ROS) production of the MLE12 cells exposed to NOX or HYX for 24 h with or without 100 µM cholesterol. Scale bars, 50 μm, n = 5/group. (C) Relative mRNA expression of Abca1, Abcg1, Ldlr, Hmgcr, normalized to GAPDH. n = 5/group. (D) Representative pictures of MLE 12 cells stained with LXRα antibody exposed to NOX or HYX for 24 h, and the quantification of the mean fluorescence intensity of LXRα. Scale bars, 50 μm, n = 5/group. The experiments were repeated at least 3 times. *P < 0.05; ** P < 0.01; *** P < 0.001
Cellular cholesterol homeostasis is primarily regulated by two key transcription factors, SREBP2 and LXR, responsible for upregulating cholesterol content and downregulating cholesterol levels, respectively [26]. We next analyzed mRNA levels of SREBP2-regulated genes and LXR-regulated genes under the hyperoxia condition, at the transcriptional level, hyperoxia exposure led to a prominent downregulation of LXR-transcribed genes including Abca1 and Abcg1 but only minor changes in SREBP2-transcribed genes (Ldlr and Hmgcr) with no significant difference (Fig. 2C). Immunofluorescence of MLE12 also showed that hyperoxia exposure decreased protein expression of LXRα (Fig. 2D), a subtype of LXR, mainly regulating the expression of ABCA1 and ABCG1 transporters, which are highly expressed in pneumocytes [27, 28].
LXR agonist attenuated hyperoxia-induced ROS production and mitochondrial dysfunctionTo evaluate the roles of the LXR pathway under hyperoxia exposure, which induces cholesterol efflux, LXR agonists GW3965 were applied. MLE12 cells were treated under hyperoxia stimulation with different doses of GW3965 for 24 h to evaluate the cell viability by a CCK8 assay. As shown in Fig. 3A, the cell viability of MLE12 cells was decreased significantly and LXR agonist GW3965 5µM exerted around a 10% increase in cell proliferation under hyperoxia exposure.
Fig. 3
The LXR agonist attenuated hyperoxia-induced mitochondrial dysfunction and reduced reactive oxygen species (ROS). (A) MLE12 cells were cultured under normoxia (NOX) or hyperoxia (HYX) conditions with different doses of GW3965 for 24 h, and cell viability was determined by CCK-8 assays. n = 4/group. (B) Relative mRNA expression of Abca1 normalized to Gapdh. n = 4/group. (C-D) Representative pictures of MLE12 cells exposed to NOX or HYX for 24 h treated with or without 5 µM GW3965 stained by Filipin III and the quantification of the mean fluorescence intensity of Filipin III staining. Scale bars,50 μm, n = 4/group. (E) Reactive oxygen species (ROS) production of the MLE12 cells exposed to NOX or HYX environment. Scale bars, 50 μm, n = 4/group. (F-G) Representative pictures of MLE 12 cells stained with JC-1 exposed to NOX or HYX for 24 h, and the quantification of the mean fluorescence intensity of JC-1. Dye accumulation in mitochondria was detected by fluorescence microscopy (aggregate red form with absorption/emission of 585/590 nm and green monomeric form with absorption/emission of 510/527 nm). The red/green fluorescence ratio of JC-1 dimers to monomers in MLE12 cells stained with JC-1. The histogram showed the red/green fluorescence ratio of JC-1 dimers to monomers in MLE12 cells stained with JC-1. Scale bars, 50 μm, n = 5/group. The experiments were repeated at least 3 times. *P < 0.05; ** P < 0.01; *** P < 0.001
Hyperoxia stimulation resulted in a decrease of approximately 70% in the mRNA level of Abca1, whereas the LXR agonist GW3965 5µM notably increased the mRNA expression of Abca1 in a dose-dependent manner (Fig. 3B). Additionally, GW3965 5µM exhibited the ability to diminish intracellular cholesterol accumulation in response to hyperoxia stimulation (Fig. 3C and D), leading to a reduction in ROS production under the hyperoxic environment (Fig. 3E). Elevated ROS levels are associated with reduced membrane potential and cell apoptosis [29]. Lower numbers of MLE 12 cells with the red dimeric form of JC-1 (indicative of normal status) in contrast to the ones with green JC-1 monomers (representing a depolarized mitochondrial membrane) were observed after hyperoxia exposure, indicating loss of mitochondrial membrane potential (Fig. 3F and G). Interestingly, this trend was reversed when treated with 5µM GW3965 (Fig. 3F and G). These findings suggested that activation of the LXR pathway can reduce hyperoxia-induced ROS production and alleviate mitochondria dysfunction by reducing cholesterol overaccumulation in MLE12 cells.
LXR agonist protected against hyperoxia-induced cell apoptosisHyperoxia increased the relative protein expression of the apoptosis-related protein BAX whereas there was a decrease in the expression of anti-apoptosis protein BCL2 in MLE12 cells exposed to hyperoxia (Fig. 4A). To investigate the effect of LXR agonist on hyperoxia-induced apoptosis in MLE12 cells, we further analyzed the alternation of the BCL2 and BAX protein expression. Results from western blot and qRT-PCR analysis revealed that LXR agonist GW3965 administration reversed the elevated BAX/GAPDH ratio associated with proapoptotic effects and amplified the reduced BCL2/GAPDH ratio linked to antiapoptotic effects triggered by hyperoxia, which may be associated with the activation of β-catenin pathway (Fig. 4A and B). In order to conduct a more thorough examination of the impact of GW3965 treatment on MLE12 cells in the presence of hyperoxia, apoptosis staining results revealed that GW3965 reduced the percentage of FITC V-positive MLE12 cells, along with the proportion of FITC V-positive /PI-positive MLE12 cells under hyperoxic stimulation, demonstrating an anti-apoptotic influence (Fig. 4C and D). These results suggested that LXR agonist inhibits hyperoxia-induced apoptosis in MLE12 cells. At the mRNA and protein level, hyperoxia lowered the expression of the epithelial cell marker SFTPC in MLE12 cells as an indicator of ATII (Fig. 4D and E). Treatment of MLE12 with GW3965 had a positive effect on the expression of SFTPC under hyperoxic stimulation (Fig. 4D and E). We also found that the increased mRNA expression of inflammatory cytokines including IL-6, and TNF-α induced by hyperoxia exposure was downregulated by GW3965 treatment (Fig. 4F).
Fig. 4
LXR agonist protected against hyperoxia-induced cell apoptosis. (A) The relative protein levels of BCL2, BAX, and β-catenin were determined by western blot analysis and quantified. n = 4/group. (B) The relative mRNA expression of Bcl2 and Bax was determined by qRT-PCR, normalized to Gapdh. n = 4/group. (C) Representative pictures of MLE12 cells stained with Annexin FITC and PI. Scale bars, 50 μm, n = 4/group. (D) The immunofluorescence staining and fluorescent quantitation of SFTPC in MLE12 cells exposed to normoxia (NOX) or hyperoxia (HYX), and HYX with GW3965 intervention. Scale bars, 50 μm, n = 4/group. (E) The relative mRNA expression of Sftpc, normalized to Gapdh. (F) The relative mRNA expression of Il-6 and Tnf-α, normalized to Gapdh. n = 4/group. The experiments were repeated at least 3 times. *P < 0.05; ** P < 0.01; *** P < 0.001
LXR agonist exhibited protective effects on lung injury in BPD rat modelTo explore whether LXR pathway plays a role in the development of BPD in vivo, we activated LXR signal pathway with LXR agonist T09 in the BPD rat model. SD newborn rats were assigned to the normoxia environment (RA group), hyperoxia with PBS (BPD + PBS group), or hyperoxia with LXR agonists (BPD + T09 group). Morphological analysis of lung tissue sections stained with HE revealed that the BPD + PBS group had higher MLI and lower RAC compared to the RA group, which was reversed by administration of T09 (Fig. 5A and B). It indicated that the LXR agonist improved alveolarization in newborn rats exposed to hyperoxia. Western blot analysis also showed that T09 supplementation increased protein levels of CD31, VEGFA (vascular endothelial cell markers), and SFTPC (markers of ATII) reduced by hyperoxia exposure (Fig. 5C).
We also observed abnormal α-smooth muscle actin (α-SMA) distribution throughout the interstitium in lung tissues of the BPD group. The increased expression of the fibrosis marker α-SMA induced by hyperoxia was reversed after T09 administration (Fig. 5C and D). Additionally, the enhanced mRNA expression of inflammatory cytokines including IL-6, TNF-α, and CXCL1 induced by hyperoxia exposure was downregulated by T09 intervention, regardless of statistical difference (Fig. 5E). Nevertheless, the LXR agonist did not appear to influence the differentiation of ATII cells into ATI cells, as there was no significant difference in aquaporin 5 gene expression after T09 intervention (Fig. 5E). There was no significant difference in body weight between the BPD + T09 treatment group and the BPD group (Supplementary Fig. 2A). These findings suggested that T09 treatment may alleviate BPD by repairing damage to ATII cells caused by high oxygen concentration exposure, potentially leading to improvement in vascular development and inflammation.
Fig. 5
LXR agonist exhibited protective effects against lung injury in BPD rat model. (A) Representative photographs of HE-stained lung sections from rats exposed to room air (RA), hyperoxia with PBS administration (BPD + PBS) and hyperoxia with T0901317 administration (BPD + T0901317 group). Scale bars, 100 μm (100X) and 50 μm (200X), n = 5/group. (B) Quantification of the HE-stained lung sections of the rats, shown as mean linear intercept (MLI) and radial alveolar counts (RAC), n = 5/group. (C) The relative protein levels of CD31, VEGFA, SFTPC, α-SMA and GAPDH were determined by western blot analysis, with quantification shown as histograms. n = 4/group. (D) Lung tissue sections were stained for α-SMA depicted in red and DAPI (shown in blue). In the RA group, the α-SMA was mainly located at the tips of the secondary crests while in the BPD group, α-SMA was extensively present throughout the interstitial spaces. Scale bar, 20 μm. n = 4/group. (E) The relative mRNA expression of Il-6, Tnf-α, Cxcl1, aquaporin 5 (Aqp5) and α-SMA, normalized to Gapdh. n = 5/group. The experiments were repeated at least 3 times. *P < 0.05; ** P < 0.01; *** P < 0.001
LXR pathway activation reduced hyperoxia-induced accumulation of lung cholesterol and alleviated oxidative stress-induced apoptosisTo further explore the role and potential mechanism of LXR activation, by using immunofluorescence staining for SFTPC, we demonstrated that hyperoxia decreased the number of SFTPC+ cells, while T09 administration significantly increased the proportion of SFTPC+ cells (Fig. 6A and B). Besides, we used TUNEL staining to determine the cell survival in ATII in each group after the LXR agonist administration. The results showed that the relative number of SFTPC+/TUNEL+ cells was significantly reduced in the BPD + T09 intervention group compared with the BPD + PBS group (Fig. 6A and B), indicating that LXR agonist administration prevented ATII from cell apoptosis in hyperoxia-induced lung injury.
It was reported that the effects of LXR agonists on apoptosis alleviation were dependent on LXRα, but Not LXRβ [30]. LXRα mainly targets the genes involved in RCT, including ABCA1 and ABCG1 transporters [28], highly expressed in pneumocytes [27, 31]. We noted a significant decrease in LXRα protein levels after hyperoxia exposure by immunofluorescence while T09 intervention increased the LXRα protein level in lung sections (Fig. 6C and D). These results were further confirmed by western blot analysis which revealed that T09 administration reversed the hyperoxia-induced increase in the BAX/GAPDH ratio and decrease in the BCL2/GAPDH ratio, which may be linked to the activation of the β-catenin pathway (Fig. 6E). We also observed that T09 administration reduced lung TC content (Fig. 6F), and significantly decreased serum TC levels (Supplementary Fig. 2B). In addition, the malondialdehyde (MDA) levels as biomarkers of oxidative stress were also noted to be diminished by T09 administration (Fig. 6G). The above data revealed that LXR agonist treatment could alleviate hyperoxia-induced lung injury, which may be partially explained by reduced lung cholesterol overaccumulation and alleviated oxidative stress.
Fig. 6
LXR agonist reduced hyperoxia-induced accumulation of lung cholesterol and alleviated alveolar type II (ATII) apoptosis. (A) Representative photographs of SFTPC (green) /TUNEL (red) /DAPI (blue) -stained lung sections from rats exposed to room air (RA), hyperoxia with PBS administration (BPD + PBS) and hyperoxia with T0901317 administration (BPD + T0901317). Scale bars, 50 μm, n = 5/group. (B) Quantification of the SFTPC + cells and SFTPC+TUNEL+ cells of the lung sections from rats. (C-D) The representative pictures of lung sections stained with LXRα antibody and the quantification of the fluorescence. Scale bars, 20 μm, n = 5/group. (E) The relative protein levels of BCL2, BAX, β-catenin, and GAPDH were determined by western blot analysis. n = 5/group. (F) The lung total cholesterol (TC) content of the three groups of rats. n = 5/group. (G) The malondialdehyde (MDA) of the three groups of rats. n=5/group. The experiments were repeated at least 3 times. *P < 0.05; ** P < 0.01; *** P < 0.001
留言 (0)