Cell culture
L02 cells (sourced from Shanghai Zhong Qiao Xin Zhou Bio Co., Ltd.) and AML12 cells (procured from Wuhan Pu-nuo-sai Life Technology Co., Ltd.) are normal human and mouse hepatocytes, respectively, commonly used as in vitro models for studying liver injury. Cells were grown in DMEM medium (Gibco, US) containing 10% fetal bovine serum (EallBio, China) and 1% penicillin–streptomycin (Gibco, US) inside a 37°C incubator with 5% CO2.
Cell survival assay
Cells were inoculated into 96-well plates. Cells were treated with 0–80 nM triptolide (Chengdu Ruifensi Biotechnology Co., Ltd, purity > 98%) for 48 h, or with 0–16 μM Ferrostatin-1 (Fer-1, Selleck Chemicals) for 49 h, or pretreated with Fer-1 for 1 h before co-treatment with triptolide for 48 h. After treatment, the solution in each well was removed, and 0.5 mg/mL MTT (Sigma, US) was added, followed by incubation in the incubator for 1 h. Subsequently, MTT was discarded, followed by the addition of dimethyl sulfoxide (DMSO) to dissolve the formazan crystals. Optical density (OD) was then quantified at a wavelength of 492 nm using a microplate reader.
Determination of iron, malondialdehyde (MDA), reduced glutathione (r-GSH)/oxidized glutathione (GSSG), total glutathione (t-GSH) and lipid peroxide (LPO)
Iron and MDA levels in hepatocytes and liver tissues, as well as r-GSH/GSSG, t-GSH, and LPO in liver tissues, were evaluated utilizing commercial assay kits (Nanjing Jiancheng Bio Co., Ltd., China) following the provided instructions.
Measurement of intracellular ROS
DCFH-DA (Beyotime, China) was used for the detection of intracellular ROS. After AML12 cells were treated according to the experimental design, they were first incubated with DAPI for 10 min for nuclear staining. Following a PBS wash, cells were incubated with DCFH-DA for 5 min. After another PBS wash, the cells were photographed using a fluorescence microscope (Olympus, Japan).
Cell transfection
The plasmids pcDNA3.1-GPX4, pcDNA3.1-Nrf2, pcDNA3.1 empty vector (served as a negative control in overexpression experiment), and pcDNA3.1-HA-ubiquitin (HA-Ub) were constructed by Hunan Youbao Biological Company. The plasmids were extracted using a plasmid extraction kit (Omega, US) in accordance with the instructions. AML12 cells underwent transfection with the plasmids utilizing lipofectamine 2000 (Invitrogen, US). Procedurally, plasmid was mixed with lipofectamine 2000 in serum and antibiotic-free DMEM medium and left to stand for 20 min. After 6 h of cell transfection, cells were maintained in complete medium for 18 h, and then exposed to TP for a further 48 h. Transfection efficiency was evaluated through western blot assay.
Cellular thermal shift assay (CETSA)
This method is commonly used to evaluate drug-target interactions. If a substance boosts a protein's thermal stability, it usually indicates that the substance may bind to the protein. Procedurally, AML12 cells received a 24 h treatment with 70 nM TP in a 37 °C constant temperature incubator. Cells were collected, rinsed by PBS, and then suspended in PBS that includes protease inhibitors phenylmethanesulfonyl fluoride (PMSF). The cell suspensions were uniformly distributed into tubes, exposed to varying temperature (room temperature, 34℃, 37℃, 40℃, 43℃, 46℃, 52℃) for 3 min, followed by cooling for an additional 3 min at room temperature. Then, cells underwent lysis by freeze–thawed three times in liquid nitrogen and a water bath, followed by centrifugation. The lysates were prepared for western blot analysis.
Protein stability and degradation analysis
Cycloheximide (CHX), a protein synthesis inhibitor, and MG132, a proteasome inhibitor, both procured from MedChemExpress, were used to explore how TP affected Nrf2 protein. Procedurally, post 24 h TP treatment, the cells were treated with 80 μg/mL CHX for 0, 1, 2, and 4 h, or with 80 μM MG132 for 1 and 2 h, followed by collection at various time points. Western blotting was conducted to assess Nrf2 protein expression.
Immunoprecipitation (IP)
To detect the impact of TP on Nrf2 ubiquitination, AML12 cells were co-transfected with pcDNA3.1-HA-Ub and pcDNA3.1-Nrf2, followed by TP treatment. The treated cells were lysed on ice for 30 min with immunoprecipitation lysate buffer that contained protease and phosphatase inhibitors. After a 15-min centrifugation at 4°C, the supernatants were obtained and subsequently incubated for an extended period at 4°C with anti-β-actin, anti-Flag or anti-HA-Ub antibodies. Following this, the samples were combinded with protein A + G magnetic beads for 1 h (P2108, Beyotime, China). Next, the magnetic beads were rinsed with cold PBS three times and denatured, before undergoing western blot analysis.
Animal experimental design
Male specific pathogen-free (SPF) C57BL/6J mice (6 weeks, 18-22g) were supplied by Hunan Slake Jingda Experimental Animal Co., Ltd. Nrf2 knockout mice (Nrf2−/−) with a C57BL6 background were kindly provided by Dr. Shilin Luo of the Second Xiangya Hospital. Mice were kept in ventilated cages with unrestricted access to food and water. Before experiments, they were adaptive feeding for one week.
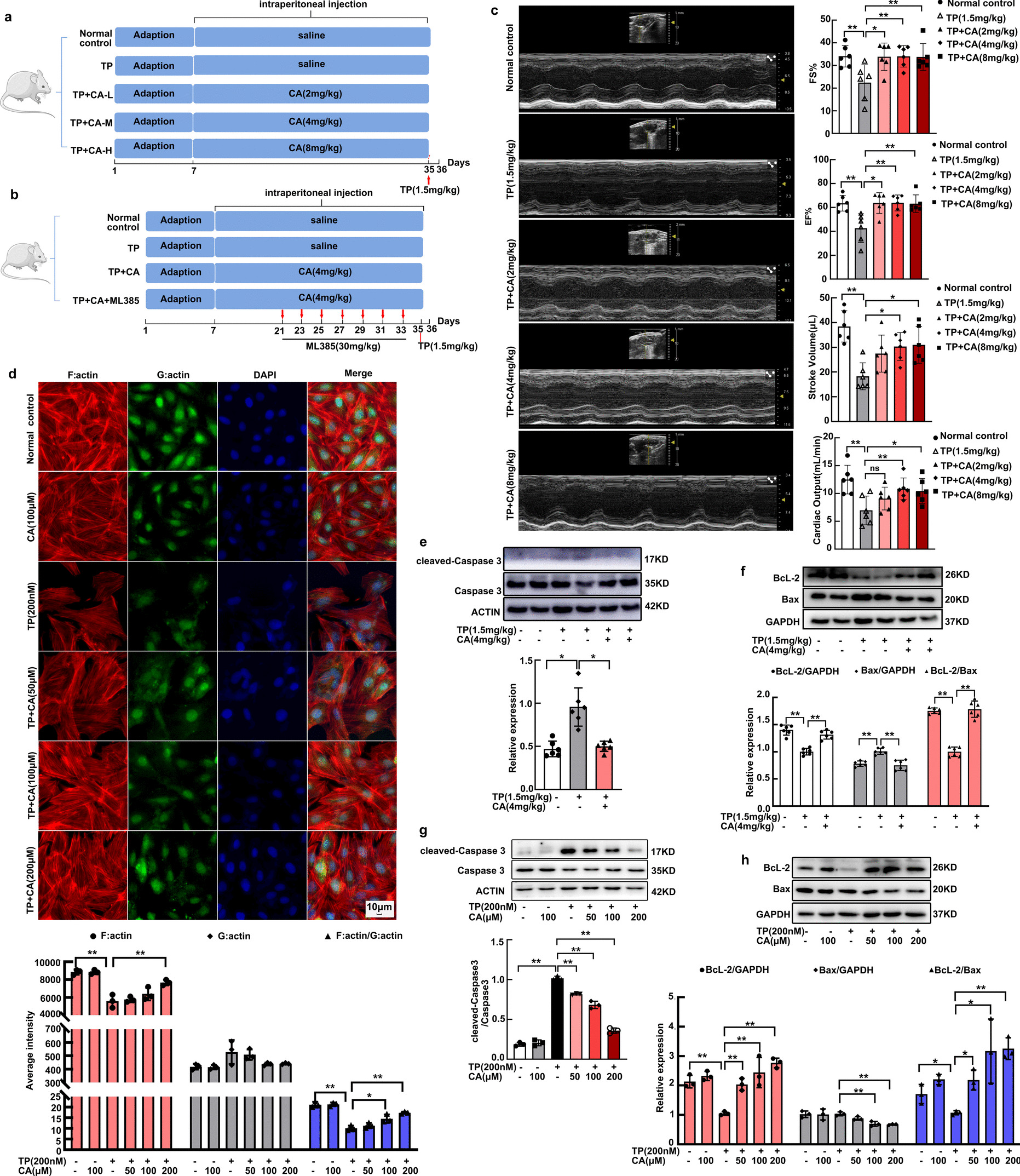
For determining the involvement of ferroptosis in TP-induced hepatic damage, C57BL/6J mice were randomly grouped into three groups (n = 8 per group): Control group, TP group, TP + Fer-1 group. TP was dissolved in physiological saline with 0.4% DMSO. Fer-1 was dissolved in a solution of 1% DMSO, 50% PEG300, 5% Tween 80, and 44% dH2O. Fer-1 (10 mg/kg/day) was given via intraperitoneal injection for 5 days before TP administration. A single dose of TP (1.2 mg/kg) was injected intraperitoneally on the 5th day. The next day, mice were anesthetized with isoflurane for blood collection and then euthanized to collect liver samples.
To study the role of Nrf2 in TP-related liver injury, Nrf2−/− mice (genotype identification in Fig. S1) were used. C57BL/6J mice were used as wild-type (WT) control. The experimental groups were designated as follows (n = 6 per group): WT group, WT + TP group, Nrf2−/− group, Nrf2−/− + TP group. The administration regimen for TP is still a single dose of 1.2 mg/kg via intraperitoneal injection.
Above mice experiments were sanctioned by the Ethics Committee of the Second Xiangya Hospital of Central South University (Approval No. 20230504).
Measurement of liver enzymes
The eyeballs of the mice were taken away for blood sampling and serum was separated via centrifugation (3,000 xg, 10 min). The serum concentrations of ALT, AST and ALP were assessed using an automatic biochemical analyzer (7600, Hitachi, Tokyo, Japan) in the Department of Clinical Laboratory of the Second Xiangya Hospital.
Hematoxylin and eosin (H&E) staining, Prussian blue staining
H&E staining of liver tissue sections was conducted to evaluate the extent of hepatic damage. Hepatic sections underwent Prussian blue staining to visualize iron deposition. Briefly, liver tissues were fixed overnight, dehydrated, embedded in paraffin, sectioned, stained with H&E or Prussian blue, and sealed. The histological images were photographed by an optical microscope (Olympus, Japan).
4-HNE immunohistochemistry (IHC)
The paraffin sections were dewaxed, followed by antigen repair and serum closure. Following that, they were exposed to primary antibodies (4-HNE, ab46545, Abcam) for incubation at 4℃. Subsequently, sections were treated with the secondary antibody, and then underwent DAB and hematoxylin staining. After dehydration and sealing, sections were observed under a microscope.
Transmission electron microscope (TEM)
The hepatic tissue was cut with the size of 1 mm3 and fixed with electron microscope fixation solution and stored at 4℃. After fixation with 1% osmic acid, the tissues were dehydrated through acetone, embedded, sliced, and stained with uranyl acetate-lead citrate. Finally, mitochondrial morphology in mouse liver was observed with a TEM (Hitachi, Tokyo, Japan).
Untargeted lipidomics analysis
Harvested livers were immediately cryopreserved in liquid nitrogen, and then placed at -80℃ thereafter. Lipids were extracted and identified by Suzhou PANOMIX Biomedical Tech Co., Ltd. (Jiangsu, China) in accordance with the published methods (Narvaez-Rivas and Zhang 2016). Briefly, the sample was added with 750 μL of chloroform:methanol (2:1, v/v. It was then ground with steel balls for 60 s and incubated on ice for 40 s. After adding 190 μL of H2O, it was vortexed and incubated on ice. Next, the mixture underwent centrifugation at 12,000 rpm for 5 s, and 300 μL of the organic phase was transferred to a new tube. This organic layer was mixed with 500 μL of solvent, and centrifuged again. Finally, the organic layer was dried, dissolved in 200 μL isopropanol and filtered for liquid chromatography-mass spectrometry (LC–MS) analysis. Chromatographic separation was accomplished using an CQUITY UPLC® BEH C18 column at 50℃, with an autosampler temperature set at 8℃. Gradient elution was performed with acetonitrile/water (60:40) (A2) and isopropanol/acetonitrile (90:10) (B2) at a flow rate of 0.25 mL/min. ESI-MSn experiments utilized a spray voltage of 3.5 kV in positive mode and 2.5 kV in negative mode. Sheath gas and auxiliary gas were set at 30 and 10 arbitrary units, respectively, with a capillary temperature of 325℃. The orbitrap analyzer performed full scans across a mass range of m/z 150–2000 at a mass resolution of 35000. Data-dependent acquisition (DDA) MS/MS experiments were conducted using HCD scan, with a normalized collision energy set at 30 eV. Normalization of the total peak area based on the signal response within samples is performed. Ropls was used for orthogonal partial least squares discriminant analysis (OPLS-DA) dimensionality reduction, and score plots were generated to show differences in lipid composition among samples. A permutation test was conducted to check for model overfitting. Statistical tests were used to calculate P values, and OPLS-DA was employed to determine variable importance in projection (VIP) and fold change for group differences. When P < 0.05 and VIP > 1, lipid molecules were regarded as statistically different.
Western blot
Cells or liver tissues were lysed and homogenized in lysis buffer (RIPA: PMSF = 100:1) for total protein extraction. The collected supernatant after centrifugation was added with 5 × SDS-PAGE loading buffer, and then heated to 99°C for a duration of 10 min. Proteins were isolated using SDS-PAGE and transferred to PVDF membranes. Following a 10-min blocking step, membranes were exposed to the primary antibodies at 4°C for 16–18 h, and to the secondary antibody for 1 h. Using an ECL kit (Bioscience, Shanghai, China), bands were visualized with a ChemiDoc Imager (BIO-RAD, US). Ultimately, their intensities were measured using Image J 1.52a. The primary antibodies employed were listed below: anti-SLC7A11 (ab175186, Abcam, 1:10,000), anti-GPX4 (BM5231, Boster, 1:2000), anti-FTH1 (DF6278, Affinity, 1:2000), anti-HO-1 (10,701–1-AP, Proteintech, 1:3000), anti-Nrf2 (AF0639, Affinity, 1:1000), anti-Flag Tag (T0003, Affinity, 1:5000), anti-HA Tag (AF0039, Beyotime, 1:1000), anti-β-actin (ac026, ABclonal, 1:10,000). The secondary antibodies utilized were: HRP-conjugated anti-rabbit IgG (BA1054, Boster, 1:5000) and HRP-anti-mouse IgG (BA1054, Boster, 1:5000).
Real-time quantitative PCR (RT-qPCR)
Total RNA was obtained using the TRIzol reagent. RNA samples were converted into cDNA by EVO M-MLV Reverse Transcription Kit (Accurate Biology, Hunan, China), starting with 1 μg RNA. The concentration and purity were evaluated applying a NanoDrop spectrophotometer (Thermo Fisher, US). RT-qPCR was conducted using SYBR Green Premix Pro Taq HS qPCR Kit (Accurate Biology, Hunan, China) on a fluorescent qPCR machine (Applied Biosystems, US). The gene expression levels were normalized against GAPDH and assessed through the 2−ΔΔCt method. Primer sequences were listed in Table S1.
Statistical analysis
Data were statistically analyzed employing SPSS 25.0 and results were presented as Mean ± SEM. Independent sample t-test was employed for two-group comparisons, and one-way analysis of variance (ANOVA) was applied for three or more groups. For pairwise comparisons, the least significant difference (LSD) test was utilized. P < 0.05 was indicated to denote statistical significance. Statistical graphs were produced employing GraphPad prism 6.0 software.




留言 (0)