Clinical samples
Peripheral blood (PB) or bone marrow (BM) samples were collected from patients diagnosed with CML, Philadelphia chromosome-positive B-cell acute lymphoblastic leukemia (BCR-ABL+ B-ALL) (see Supplemental Table S1), as well as from HIs. Samples were collected from the First Affiliated Hospital of Jinan University. Written informed consent was obtained from all participants before blood collection, and the study was approved by the ethical committee of the First Affiliated Hospital of Jinan University (No.2014006).
Cell culture
Mouse 32D cells were purchased from the China Center for Type Culture Collection (CCTCC). The human CML cell line KBM5, K562-R, and 32D-BCR-ABL were generously provided by Prof. Jingxuan Pan, Sun Yat-Sen University. Other hematological malignancy cell lines including U266, RPMI8226, H929, CCRF-CEM, Molt4, Jurkat, NB4, MOLM-13, KG1-α, HL-60, MV-411, K562, K562-R, Ku-812, TOM-1, Kasumi-1, SUP-B15 and 293T cells were provided by the Institute of Hematology, Jinan University. SUP-B15, U266, RPMI8226, H929, CCRF-CEM, Molt4, Jurkat, NB4, MOLM-13, KG1-α, Kasumi-1, HL-60, MV-411, K562, Ku-812, TOM-1 were cultured in 90% RPMI 1640 with 10% Gibco fetal bovine serum (FBS). K562-R cells were cultured with 5µM IM under the same conditions. KBM5, and 32D-BCR-ABL were cultured in 90% Iscove’s Modified Dulbecco’s Medium (IMDM) with 10% FBS. 293T cells were cultured in 90% DMEM with FBS. All cell lines were maintained in a humidified atmosphere containing 5% CO2 at 37 °C. Plasmocin prophylactic was added to all media to prevent potential mycoplasma contamination.
CD34 + cells were isolated using the CD34 Microbeads Kit (#130-046-702, Miltenyi Biotec, Germany) according to the manufacturer’s instructions and cultured in IMDM supplemented with 10% FBS along with SCF (#300-07-500, Proteintech; 100 ng/mL), IL-3 (#213-13-100, Proteintech; 20 ng/mL), IL-6 (#216-16-100, Proteintech, 20 ng/mL), and GM-CSF (#300-03-100, Proteintech; 100 ng/mL) at 37℃ in a humidified incubator with 5% CO2.
Reagents
Disulfiram (DSF; S1680), Imatinib (IM; S1026), Dasatinib (DAS; S1021), Z-VAD-FMK (S7023), N-acetylcysteine (NAC; S1623), Ferrostatin-1 (Fer-1; S7243), Deferoxamine (DFO; S5742), MG132 (S2619), Chloroquine (CQ; S6999), Cycloheximide (CHX; C7698), Vitamin E (Vit-E; S4686), VX-765 (S2228), MG132 (S2619) and Necrosulfonamide (NSA; S8251) were purchased from Selleck Chemicals (Houston, TX, USA). 4-Diethylaminobenzaldehyde (DEAB; HY-W016645) was purchased from MedChemExpress (Monmouth Junction, NJ, USA). Z-VAD-FMK, Fer-1, DFO, Vit-E, NSA were dissolved in DMSO and added to culture media at a final concentration of 10 µM. NAC was dissolved in DMSO and added to culture media at a final concentration of 1 µM.
RNA interference
KBM5 cells were transfected using the Neon® Transfection System (Invitrogen) with 100 pmol of oligonucleotides in 10 µl reactions [25]. Briefly, 2 × 105 cells were suspended in 100 pmol of siHSPA8, siALDHA1, or siGPX4 in 10 µl reactions. After electroporation, cells were cultured in IMDM medium containing 10% FBS at 37 °C with 5% CO2. The transfected cells were used for subsequent experiments after 24–48 h. siRNA sequences targeting HSPA8, STUB1, BCR-ABL and GPX4/Gpx4 are listed in Supplemental Table S2.
In vivo siRNA delivery
The preparation of amine-terminated, generation 5 polyamidoamine (G5-PAMAM, hereafter called G5) dendrimer-siRNA nanoparticles for use in mouse models was conducted according to the protocol established by Prof. Daoguang Yan from Jinan University. Briefly, in vivo siRNA delivery was conducted using G5 (CYD-150 A), obtained from Weihai Chenyuan Molecular New Materials Co., Ltd. The N: P ratio of G5 to siRNA was set at 30:1, and the complexes were formed in PBS at room temperature with gentle vortexing for 10 min. A dosage of 1.0 mg/kg of the G5-siRNA complex was administered via tail vein injection every 3 days.
Establishment of stable cell lines
A total of 3 µg of the PLKO-puro-NC or PLKO-puro-sh-GPX4 plasmid along with 1.5 µg of psPAX2 and 1.5 µg of pMD2.G were transfected into 293T cells using lipofectamine 3000 (Life Technologies, USA). Virus supernatant was harvested 24 and 48 h after transfection. Subsequently, KBM5 cells and mouse lineage- cells were co-cultured with virus supernatant. Then 5 µg/mL puromycin was applied to select positively infected cells for at least 10 days. shRNA sequences targeting GPX4/Gpx4 are listed in Supplemental Table S2. Lentiviruses overexpressing HSPA8 (LV-HSPA8, GV705), GPX4 (LV-GPX4, GOSL0438191), control lentivirus (LV-NC, CON525), Flag-tagged GPX4-WT (wild type), and Flag-tagged seven cysteines-mutated GPX4 (GPX4-7CA mutant) were purchased from Genechem (Shanghai, China). KBM5 cells were transduced with LV-HSPA8 or LV-GPX4 lentivirus, and positively transduced cells were selected by treatment with 5 µg/mL puromycin for a duration of 10 days.
Cell viability assay
Cell viability was assessed using the Cell Counting Kit-8 (CCK-8) (#96992, Sigma) following the manufacturer’s protocol. In brief, cancer cell lines were seeded at 10,000 cells per well, and primary cells at 50,000 cells per well, in 96-well plates. After treatment, 10 µl of CCK-8 reagent was added to each well and incubated for 4 h. The absorbance was then measured using a microplate reader (Biotek Synergy4, USA).
Colony forming cells (CFC) assays
GFP + lineage- c-Kit + cells sorted from BCR-ABL mice were plated in M3434 methylcellulose medium (Stem Cell Technologies). Cultures were incubated at 37 °C in a humidified atmosphere of 5% CO2 for 10–14 days. For CFC assays using BCR-ABL+ leukemia cell lines, 1,000 cells were seeded into 24-well plates after a 24-hour drug treatment. Subsequently, the cells were cultured in IMDM supplemented with 2% FBS and Methylcellulose Stock Solution (HSC001, Bio-Techne). Cultures were incubated at 37 °C in a humidified atmosphere of 5% CO2 for 10 to 14 days before counting.
Lipid peroxidation and cell death analysis
BCR-ABL+ leukemia cell lines, primary cells from patients or mouse primary cells were harvested and washed three times in PBS following various treatments. Lipid peroxidation and cell death analysis were using BODIPY-C11(D3861, Invitrogen) and Annexin-V-APC/PI kit (AP107-100, MultiSciences, China), and analyzed by flow cytometry.
The labile iron pool assay
The FeRhoNox-1 probe (MX4558, MKBio, China) was used to assess the labile iron pool. Briefly, a 500 µl cell suspension from each treatment group was seeded into triplicate wells of a 24-well plate. Subsequently, 2.5 µM FeRhoNox-1 probe was added to each well and incubated for 20 min at 37 °C. Flow cytometry was then conducted for analysis.
Malondialdehyde (MDA) assay
The relative concentration of malondialdehyde (MDA), an end product of lipid peroxidation, was measured using a Lipid Peroxidation Assay Kit (S0131M, Beyotime, China) following the manufacturer’s instructions.
GSH and GSSG detection
The GSH and GSSG levels of BCR-ABL+ leukemia cell lines or tissue from mice were measured using a GSH and GSSG Assay Kit (S0053, Beyotime, China) according to the manufacturer’s instructions. The total GSH and GSSG concentrations were calculated using a standard curve and normalized to the total protein level in each sample.
Immunofluorescence analysis
Cells were washed with cold phosphate-buffered saline (PBS), fixed in 4% paraformaldehyde for 15 min at room temperature, and permeabilized with 0.1% triton X100 for 10 min at room temperature. Cells were stained with GPX4 antibody (Santa Cruz, sc-166570; 1:100) or HSPA8 antibody (ab51052, Abcam; 1:200) at room temperature for 1 h. After washing 3 times with PBS, cells were incubated with the secondary antibody of Alexa Fluor 555 labeled goat anti-rabbit IgG or Alexa Fluor 488 labeled goat anti-mouse IgG at room temperature for 30 min. 4′,6-Diamidine-2′-phenylindole dihydrochloride (DAPI) (Thermo Fisher Scientific) was used to stain the nucleus. MitoTracker Red (M7512, Invirogen) was used to stain mitochondria. Images were captured using a Leica SP8 confocal microscope (Leica Corp, Germany).
Western blot analysis
Cells were harvested and lysed in RIPA buffer supplemented with protease inhibitors. Proteins were then separated using a 12% SDS-PAGE Criterion X-gel (Bio-Rad) and transferred to a PVDF membrane (Bio-Rad). The membranes were blocked with QuickBlock™ Blocking Buffer (Beyotime, China) and incubated overnight at 4 °C with primary antibodies against GPX4 (#59735S, Cell Signaling Technology; ab125066, Abcam; 1:1000), HSPA8 (#8444, Cell Signaling Technology; 1:1000), Ubiquitin (ab134953, Abcam; 1:2000), and Flag (AF0036, Beyotime; 1:1000). The anti-β-actin (ACTB) antibody (A3853, SIGMA; 1:5000) was used as a control. Bands were visualized using enhanced chemiluminescence (ECL; Beyotime, China) and the UVITEC photo documenter.
Coimmunoprecipitation (CO-IP)
Cell protein extracts were isolated using Pierce IP Lysis Buffer (Thermo Fisher Scientific), then, cell lysates were incubated specific antibody (Anti-GPX4, sc-166570, Santa Cruz, USA) at 4 °C overnight. Dynabeads Protein A/G beads (CXB-PA/G0004-1, EeasunBio, Guangzhou) were incubated with the obtained protein complex for 6 h at 4 °C. The magnetic bead-antibody-antigen complex was washed three times with IP buffer, then eluted with loading buffer and heated at 100 °C for 10 min. The samples were resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and subsequently analyzed by immunoblotting.
RNA extraction and quantitative real-time RT-PCR
Total RNA from BCR-ABL+ cell lines or primary cells was extracted with Trizol reagent (Invitrogen) according to the manufacturer’s instructions. RNA was reverse transcribed into cDNA using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, CA, USA). Real-time PCR was conducted using SYBR Green (Tiangen, China), following a cycling program of 45 cycles at 95 °C for 30 s, 60 °C for 30 s, and 72 °C for 30 s. Relative expression levels were determined using the comparative 2-ΔΔCt method, with data normalized to ACTB mRNA levels. The primers used for real-time PCR analysis are listed in Supplemental Table S3.
RNA-seq analysis
KBM5 cells were treated with DMSO (Ctrl) or 0.25 µM DSF, both in the absence and presence of 0.25 µM IM for 48 h. Total RNA was extracted and assessed for RNA integrity using the RNA Integrity Number (RIN). cDNA library construction and sequencing were performed by Shanghai Biotechnology Corporation using the VAHTS Stranded mRNA-seq Library Prep Kit for Illumina®. High-quality reads were aligned and mapped to the human reference genome (GRCh38) using Hisat2 (version 2.0.4). Gene expression levels were calculated in terms of fragments per kilobase of exon model per million mapped reads (FPKM) and are provided in Supplemental Table S4.
Flow cytometric analysis
BM and spleen cells harvested from recipient mice were analyzed by flow cytometry (BD LSRFortessa) following staining with the specified antibodies. For the detection of leukemia myeloid cells, cells were stained with anti-mouse Gr-1-APC (E-AB-F1120UE, Elabscience), anti-mouse Mac-1-PE (E-AB-F1081UD, Elabscience), or anti-human CD45-FITC (#368508, BioLegend). To identify lineage-negative Sca-1 + c-Kit+ (LSK) cells, cells were stained with lineage-PerCP-Cy5.5 (#561317, BD), Sca-1-APC (160904, BioLegend), and c-Kit-BV421 (#567818, BD). For the identification of long-term hematopoietic stem cells (LT-HSCs) and short-term hematopoietic stem cells (ST-HSCs), cells were stained with lineage-PerCP-Cy5.5 (#558074, BD), Sca-1-APC (#17-5981-82, eBioscience), c-Kit-BV421 (#105828, BioLegend), CD135-PE (#12-1351-83, eBioscience), CD150-PE-Cyanine7 (#25-1502-82, eBioscience), and CD48-APC-eFluor780 (#47-0481-82, Invitrogen). The gating strategies used for flow cytometry analysis are detailed in Supplemental Fig. S1A.
Molecular docking
The molecular structures necessary for docking studies were obtained from the Protein Data Bank (https://www.rcsb.org/) and AlphaFold (https://alphafold.ebi.ac.uk/). For molecular docking predictions on the Windows platform, AutoDock 4.2.6 and PyMOL 2.5.4 were employed, while AutoDock Vina and GRAMM (Global RAnge Molecular Matching, https://gramm.compbio.ku.edu/) were utilized on the Ubuntu system.
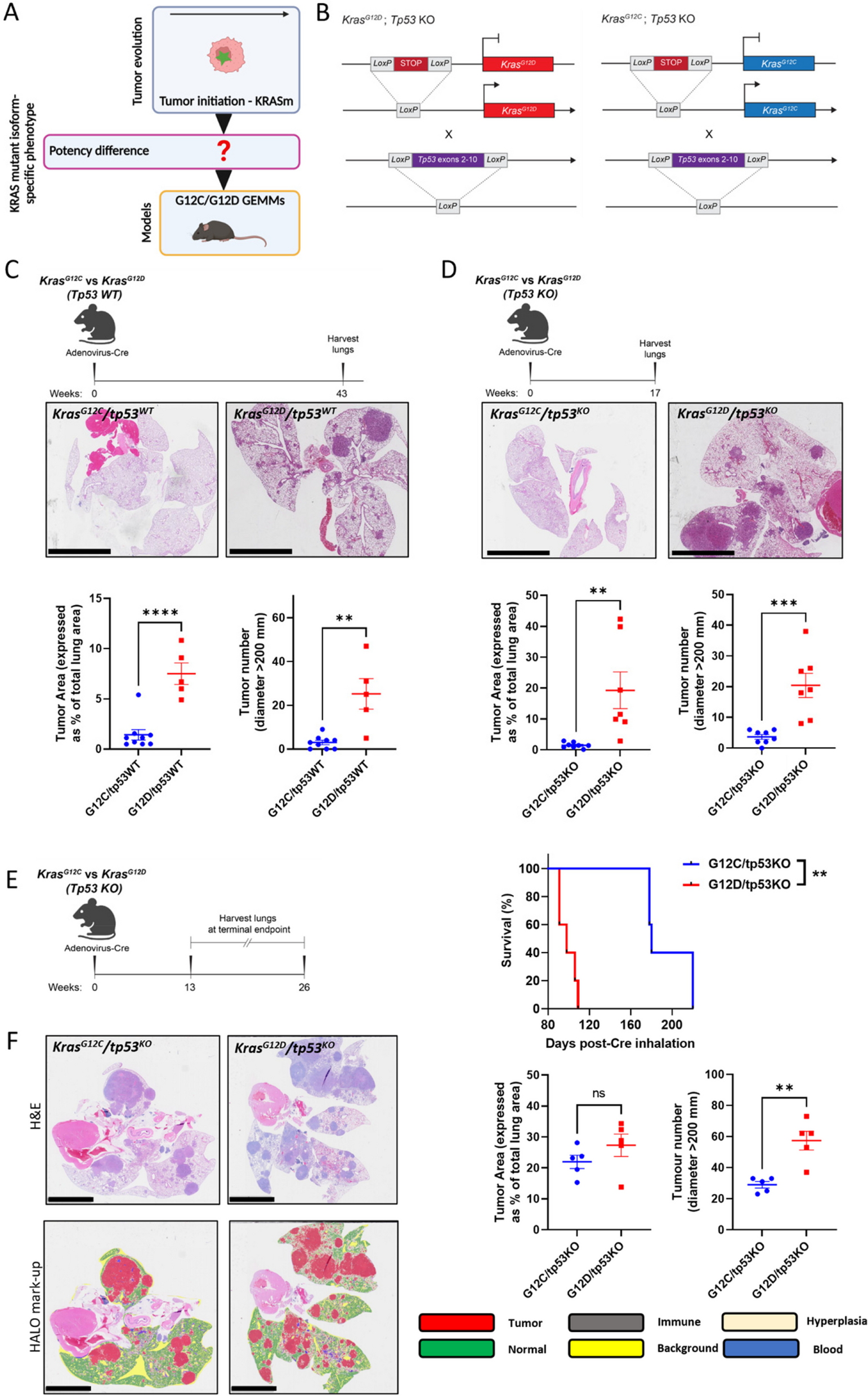
KBM5 mice
sh-NC or sh-GPX4 KBM5 cells (2 × 106 cells/mouse) were transplanted into B-NDG mice via tail vein injection to establish CML-CDX mouse models (n = 8). B-NDG mice were procured from Biocytogen (Beijing, China). Seven days post-administration of KBM5 cells, flow cytometry using a BD LSRFortessa was employed to assess the expression of human CD45 + cells. The mice were subsequently divided into three groups: sh-NC, sh-GPX4-1, and sh-GPX4-2. At day 28, three mice were randomly selected from each group, and the weight and size of the spleen (n = 3) were recorded. Tumor burden in the spleen and bone marrow of each group (n = 3) was assessed, and the survival times of the remaining mice in each group (n = 5) were documented. The animal experimental protocols were approved by the Laboratory Animal Ethics Committee of Jinan University (Approval No. 20230812-12).
K562 mice
K562 cells stably expressing both GFP and luciferase (K562-GL, 5 × 106 cells/mouse, IV) were transplanted into B-NDG mice via tail vein injection to establish CML-CDX mouse models (n = 5). B-NDG mice were obtained from Biocytogen (Beijing, China). Bioluminescence imaging was performed using the IVIS imaging system one week post-injection of K562 cells, following luciferin administration. The mice were then randomly assigned to one of four treatment groups: a control group receiving vehicle, an IM monotherapy group (100 mg/kg/day), a DSF monotherapy group (100 mg/kg/day), and a combined treatment group (100 mg/kg/day IM + 100 mg/kg/day DSF). Tumor burden progression was monitored weekly via bioluminescence imaging, and the mice were observed throughout the study period to evaluate survival outcomes. The animal experimental protocols were approved by the Laboratory Animal Ethics Committee of Jinan University (Approval No. 20211109-19).
BCR-ABL mice
The Scl-tTa-BCR-ABL1/GFP (hereafter referred to as BCR-ABL) mice were provided by Prof. Weizhang Wang, Guangdong Pharmaceutical University. FVB-N mice were purchased from HFK Bio-Technology (Beijing, China). To evaluate the therapeutic potential of DSF and TKIs, BM cells were harvested from BCR-ABL mice after a 4-week induction of BCR-ABL expression via tetracycline withdrawal. BM GFP + cells (1 × 106 cells/mouse, IV) were transplanted into lethally irradiated wild-type FVB/N recipient mice, which received 900 cGy of radiation. Blood samples were collected 4 weeks post-transplantation to confirm the onset of leukemia. The mice were then subjected to various treatment regimens: IM (100 mg/kg/day, oral gavage), DSF (100 mg/kg/day, oral gavage), a combination of DSF and IM, or a control group receiving only a vehicle (0.5% CMC-NA). After 4 weeks of treatment, a subset of animals from each cohort was euthanized for post-treatment analysis, while the remaining mice were monitored for survival. To assess the impact of DSF and TKIs treatment on LSC burden, 1 × 106 BM GFP + cells from treated mice were transplanted into secondary (2nd) recipient mice, and engraftment rates were monitored.
To evaluate the antileukemic efficacy of GPX4 deletion in maintaining BCR-ABL+ leukemia, 1 × 106 BM GFP + cells from 4-week Tet-off BCR-ABL mice were transplanted into lethally irradiated primary recipients. These mice were divided into two groups and treated with G5-siNC or G5-siGpx4 for 2 weeks. Circulating blasts and LSKs were assessed, and survival outcomes were compared between the two groups. To further investigate the antileukemic efficacy of GPX4 deletion during leukemogenesis, 5 × 105 preleukemic lineage-c-Kit + cells were sorted and transplanted into lethally irradiated recipient mice. BCR-ABL mice received G5-siGpx4 (1 mg/kg/day, IV) or G5-siNC (1 mg/kg/day, IV) for 4 weeks, starting the day after transplantation and Tet-off BCR-ABL induction. Circulating blasts, LSKs, and survival were compared between the two groups. The animal experimental protocols were approved by the Laboratory Animal Ethics Committee of Jinan University (Approval Nos. 20230313-06, 20230602-13, 20230920-01, and 20240709-01).
Statistical analysis
All statistical analyses were conducted in R language software (version 3.8.2, https://www.r-project.org/) and GraphPad Prism (version 8.0, CA, USA), as appropriate. Differences in two independent subgroups in clinical samples were tested by Mann-Whitney-Wilcoxon. For comparisons between two groups in vivo and in vitro experiments, two-tailed unpaired or paired Student’s t test was utilized. For comparisons among three or more groups, a one-way ANOVA followed by Bonferroni’s post hoc test was used. To compare a control group with multiple experimental groups, a one-way ANOVA with Dunnett’s post hoc test was applied, unless otherwise specified. The “surv_cutpoint” function in the “survminer” package was used to determine the optimal cut-off values of GPX4. Kaplan-Meier curves were compared by log-rank test using the R package “survival”. The differentially expressed genes in RNA-seq data were identified utilizing the package “edgeR”. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment were analyzed by the packages “clusterProfiler” and “org.HS.eg.db”. A two-tailed P value < 0.05 was considered statistically significant.


留言 (0)