
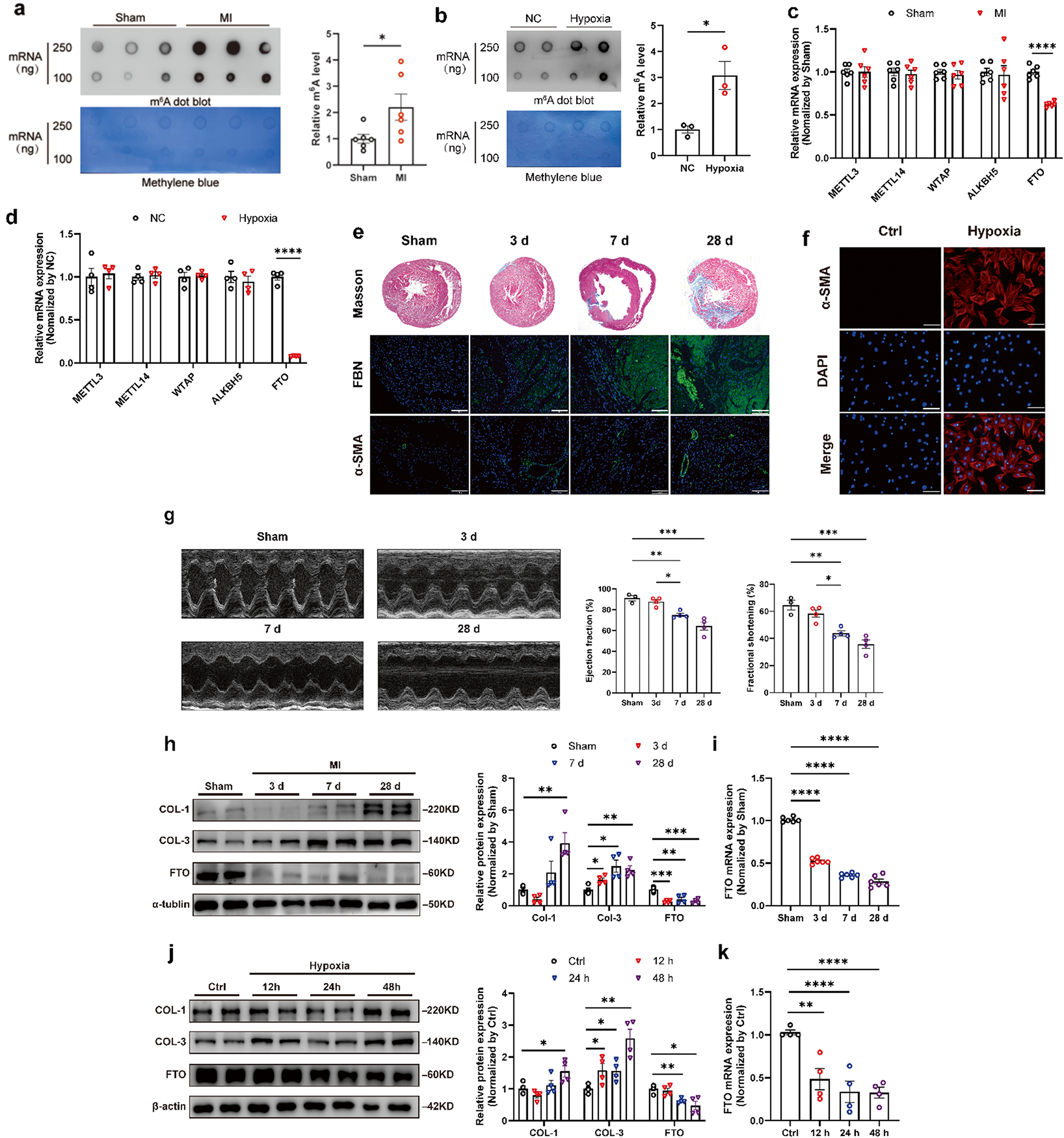
記住我



To unravel the complex changes within the ESCC microenvironment, we utilized single-cell RNA-sequencing data from patient-derived samples, obtained from GSE160269 in GEO database. (Zhang et al. 2021) Detailed analysis of 44,206 cells from both cancerous and surrounding non-cancerous tissues facilitated the identification of six primary cell types: epithelial cells (AOC1), endothelial cells (PECAM1), fibroblasts (PDGFRA), myeloid immune cells (CD14), T cells (CD3D), and B cells (CD39) (Fig. 1A–C). We confirmed these cell populations through the evaluation of known marker genes, confirming the accuracy of our cellular classification (Fig. 1B–D).
Fig. 1
Spectrum of Cell Types Detected through scRNA-Seq Analysis. A UMAP plot illustrating the single-cell data distribution across different cell types in ESCC tissues. B Violin plot depicting the expression levels of classic marker genes across various cell types. C UMAP plots demonstrate the expression patterns of specific marker genes for each cell type. The color gradient from gray to purple indicates increasing levels of gene expression. D Heatmap displaying the expression levels of the top 50 marker genes for each cell type, with functional annotations on the right. The color gradient from purple to red denotes lower to higher expression levels. E Box plots compare cell type proportions between healthy and tumor tissues, showing differences across epithelial cells, endothelial cells, fibroblast cells, myeloid cells, T cells, and B cells
Our investigation showed a significant increase in the proportion of Epi (epithelial cells) in the tumor environment (Fig. 1E), suggesting tumor growth and a proliferative index, which mirror the malignancy’s stage and capacity for expansion. Additionally, the rise in Endo (endothelial cells) number indicates an ongoing angiogenic process, essential for tumor sustenance and development. This enhancement of the tumor’s vascular network not only improves the supply of nutrients and oxygen but may also establish pathways for metastatic spread.
The tumor stroma displayed significant fibrotic features, evidenced by a heightened presence of Fib (fibroblasts) (Fig. 1E). This increase suggests a role for these cells in altering the structural and functional aspects of the tumor environment, potentially affecting the cancer's invasiveness and responsiveness to treatment.
A particularly troubling finding was the decrease in T cell proportions within the tumor niche (Fig. 1E), a critical element of the anti-tumor immune response. This decrease hints at an adept evasion of immune surveillance by the tumor, potentially allowing for unchecked tumor progression and underscoring the necessity for a better understanding of the immune landscape in ESCC.
Collectively, the data illuminate the complex cellular interactions within the ESCC microenvironment, emphasizing the roles of proliferative epithelial cells, angiogenic endothelial cells, and fibrogenic fibroblasts, set against the backdrop of a diminished T cell-mediated immune response. These insights not only enhance our comprehension of the biological mechanisms of ESCC but also provide a basis for t creating targeted therapies that focus on these specific cellular activities.
Exploring transcriptomic changes across cell types during ESCC developmentOur thorough analysis of gene expression changes across different cell types during the progression of ESCC uncovered extensive transcriptional shifts, with 1482 genes exhibiting increased expression and 1369 showing a decrease. This marked reconfiguration of the genetic framework highlights the cellular adjustments to oncogenic transformation (Fig. 2A–D).
Fig. 2
Alterations in the transcriptional landscapes of various cell types throughout tumor genesis. A Left: Heatmap displaying genes upregulated in tumor samples relative to healthy controls across various cell types. Right: Dot plot depicting the functional annotations associated with these upregulated genes. B Left: Heatmap illustrating genes downregulated in tumor samples compared to healthy controls for each cell type. Right: Dot plot showing the functional annotations of these downregulated genes. C Heatmap showing the top 100 upregulated DEGs during tumor genesis. D Heatmap showing the top 100 upregulated DEGs during tumor genesis. E Interaction network within the ESCC tumor microenvironment, highlighting increased (red lines) and decreased (blue lines) interactions among cell types compared to normal tissue.F: Bar graph showing the interaction flow among signaling pathways in tumor (orange) and normal (green) tissues, with pathways ordered by their proportional intensity in the tumor environment. G–N Heatmaps depicting the log2-fold change of SPP1 (G), TGF-beta (H), MIF (I), collagen (J), MHC I (K), CXCL (L), NOTCH (M), TNF (N) signaling across various cell types in tumor tissue relative to normal, with the color gradient shifting from red (indicating upregulation) to blue (indicating downregulation)
Upon analyzing genes with heightened expression, a distinct pattern emerged underscores the activation of genes associated with fibrotic activities, particularly noted in the enrichment of “Extracellular matrix” and “Signaling by TGF-beta family members”, as well as genes that facilitate “Epithelial to mesenchymal transition (EMT)”. These upregulated pathways indicate a strengthened fibrotic environment within cancer tissues. The increased expression of EMT-related genes highlights crucial morphological alterations in epithelial cells that are essential for cancer development. Furthermore, a significant rise in “Canonical glycolysis” observed in epithelial and endothelial cells points to a metabolic shift that supports cancer progression. Specifically, endothelial cells exhibited a notable increase in angiogenic genes, including “Angiogenesis” and “VEGFA-VEGFR2 signaling”, suggesting active vascular remodeling to accommodate tumor growth. The elevated gene expression in immune cells, notably T cells and B cells, in “Response to tumor cell” and “Positive regulation of tumor necrosis factor production” pathways, indicates a vigorous immune response to cancer cells (Fig. 2A, C).
Conversely, the noted decrease in gene expression linked to “Cell surface pattern recognition” in both epithelial and endothelial cells might indicate strategic immune evasion tactics by the tumor. A metabolic shift was also suggested by the reduction in “Oxidative phosphorylation”, which may promote reliance on glycolytic pathways. Furthermore, the lowered expression of genes essential for “Apoptosis”, “DNA repair”, and “Chromatin organization” hints at potential survival strategies that support malignant cell growth. Additionally, the diminished expression in immune-related pathways, such as “Antigen receptor-mediated signaling pathway” and “MHC class I protein binding”, raises concerns regarding the immune system's capacity to effectively target and eradicate cancer cells (Fig. 2B, D).
The detected gene expression dynamics underscore the transformative condition of the ESCC microenvironment. Increased gene expression in fibroblasts, epithelial, and endothelial cells indicates an adaptive response leading to fibrotic and metabolic modifications. On the other hand, the reduction in specific genes within endothelial, epithelial, and T cells might represent a strategic adjustment by these cells in the tumor setting. Altogether, these variations depict a complex and detailed cellular reaction within the ESCC framework, emphasizing the necessity for a comprehensive understanding of the molecular and cellular mechanisms involved in the advancement of ESCC.
Comprehensive analysis of cell-cell communication and pathway dynamics in the tumor microenvironmentIn our efforts to further clarify the changes in the tumor microenvironment (TME) during tumorigenesis, we explored the detailed cell-cell communication mechanisms among various cell types. We noted a significant increase in the interactions between Epi, Endo, and Fib throughout the tumor development. This escalation highlights the mutual reinforcement between tumor tissues and the adjacent neovasculature, as well as an increased level of fibrosis. Contrarily, immune cells, particularly TC and BC, exhibited a notable decrease in their interactions with other cellular components (Fig. 2E). This reduction further suggests the tumor cells' proficiency in evading immune detection. Overall, our observations emphasize crucial changes in cell-cell communications, indicating a combined enhancement of tumor progression and angiogenesis, alongside a reduced engagement of immune-cell within the TME.
From a broader perspective, our cell-cell communication analysis revealed significant alterations in various pathways within the TME. Key among these were notable upregulations in the SPP1, TGFbeta, MIF, COLLAGEN, and CD45 pathways, contrasted with distinct downregulation in the TNF, MCHI & MHCII, CXCL, CDH5, and NOTCH pathways (Fig. 2F). These modifications shed light on the subtle environmental changes that transpire as the tumor develops.
Exploring cell-specific pathway activities further, we noted significant upregulation of the SPP1 pathway, particularly in Epi, Endo, Fib, and Mye (myeloid immune cells). Its widespread elevation across these cell types highlights its essential role in facilitating diverse interactions within the TME (Fig. 2G). Both the TGF-beta and MIF pathways exhibited notable upregulation in Epi, Endo and Fib cells, indicating their combined impact on cellular growth, matrix alteration, and immune evasion within the tumor environment (Fig. 2H, I). Specifically, The COLLAGEN pathway was prominent in Fib cells as they engaged with other cells, emphasizing the fibroblasts’ primary role in adjusting the extracellular matrix, which may affect tumor stiffness and migration (Fig. 2J).
Conversely, the MHC and CXCL pathways showed a significant decrease in activity, particularly in the interactions between TC and BC with other cellular entities (Fig. 2K, L), potentially aiding immune evasion and giving tumor cells a hidden advantage in the TME. This noticeable reduction may be a strategic modification by tumor cells to avoid detection and suppression by the immune system. The NOTCH pathway displayed dual behavior, with upregulation in EC enhancing angiogenesis, thus encouraging the formation of new blood vessels. However, its downregulation in Epi cells suggests a change in epithelial tissue connectivity, marking a transformation towards tumorigenic traits (Fig. 2M). The TNF pathway, crucial for inflammatory responses, was upregulated in interactions among immune cells, reflecting the body's intensified response to the tumor, indicative of efforts to inhibit its growth. Nevertheless, its downregulation in Epi and Endo cells may signify the tumo’s advancement, reflecting the adaptive strategies the tumor employs to ensure its survival and progression (Fig. 2N).
In summary, our research offers a detailed view of the dynamic interactions among various signaling pathways and their cell-specific behaviors. The varied regulations across these pathways not only underscoret the tumor's adaptive strategies but also illuminate the changing communications within the TME, potentially providing insights for therapeutic interventions.
Tracing epithelial cell transformations and gene expression patterns in ESCC developmentIn our detailed exploration of the specific changes that epithelial cells experience during ESCC development, we applied pseudotime analysis to map out the different cellular states across the carcinogenic progression of these cells. Notably, our analysis delineated three unique cellular states, each shedding light on various behaviors of epithelial cells throughout ESCC progression: "Cell state 1" was present in both normal and ESCC tissues, likely indicative of typical epithelial cells. "Cell state 2," primarily found in normal tissues, seemed to represent the normal differentiation pathway of epithelial cells. Conversely, "Cell state 3," mainly observed in ESCC tissues, appeared to mark the onset of malignant transformation in epithelial cells (Fig. 3A, B). The progression from "cell state 1" to "cell state 2" reflects standard physiological differentiation, whereas the transition to "cell state 3" suggests the pathological shift from normal to cancerous states, underscoring the intricate evolution of epithelial cells from normality to malignancy in ESCC.
Fig. 3
The cellular and molecular transformations of epithelial cells across tumor progression. A Pseudotime trajectory plot of of epithelial cells in ESCC. Left, pseudotime sequence scores of epithelial cells. Top right, the distribution of epithelial cells in healthy group. Bottom right, the distribution of epithelial cells in tumor group. B Ridge plot showing the cell proportion distribution of healthy and tumor epithelial cells along pseudotime trajectory of A. C Heatmap showing the time-related gene expression profiles during tumor genesis, with gene function annotation on the right. D Ridge plots showing the expression score of gene set from different clusters in C of healthy and tumor groups. E Scatter plots and trajectory plots illustrating the expression levels of key genes identified in C
In our efforts to decode the dynamic gene expression shifts during ESCC development, we analyzed gene expression variations along two distinct differentiation trajectories, identifying two pivotal gene clusters with specific roles in the transformation of epithelial cells in ESCC (Fig. 3C, D).
Cluster 1 comprises genes that show a decrease in expression within the ESCC environment while either maintaining or increasing expression during normal differentiation. This cluster contains genes critical for for ESCC progression, including those linked to the Hippo pathway, whose downregulation suggests an ESCC-induced subversion of growth control and tissue structure. Additionally, genes involved in Oxidative phosphorylation are reduced, indicative of a metabolic shift towards glycolysis, essential for cancer cell proliferation in ESCC. The regulation of proteolysis gene group indicates a potential ESCC-induced disturbance in proteostasis, possibly enhancing metastatic potential. A reduction in genes essential for cellular homeostasis marks a disruption in internal balance, crucial in ESCC development. Furthermore, the reduction of apoptosis-associated genes points to an ESCC- promoted avoidance of cell death, contributing to cellular immortality.
Conversely, Cluster 2 encompasses genes that are upregulated during ESCC development, featuring genes crucial to oncogenesis. This includes the PI3K-Akt signaling pathway, a key conduit for cell survival and proliferation, whose upregulation suggests a heightened potential for ESCC progression. The increase in genes associated with signaling by TGFβ family members highlights enhanced interactions with TGF-β, a major regulator of cell fate and oncogenesis. An increase in genes facilitating the epithelial to mesenchymal transition illustrates the vital phenotypic shift that equips epithelial cells with invasive and migratory traits, fundamental to ESCC metastasis. The heightened expression of canonical glycolysis genes reflects a metabolic shift towards a glycolytic phenotype, a strategic adaptation of ESCC cells. Additionally, the rise in genes related to signaling by ALK in cancer highlights a possible oncogenic signaling hub within ESCC.
In the complex landscape of ESCC, our focused gene expression analysis has pinpointed crucial genes that mark the transition from normalcy to malignancy (Fig. 3E). The genes STAT3, HIF1A, and TGFB3 emerge as upregulated markers in the tumorigenic process, maintaining stability in non-malignant tissues. STAT3, with its varied roles in transcriptional regulation, seems integral to ESCC progression, potentially guiding cellular proliferation and immune evasion. The notable rise in HIF1A expression, key in regulating responses to low oxygen levels, highlights its likely role in neovascularization and the aggressive behavior of tumor cells in the hypoxic areas of ESCC. Furthermore, TGFB3's involvement in the TGF-β signaling pathway suggests its role in altering immune responses and fostering oncogenic growth. In stark contrast, the genes LHPP, HMOX1, and BCL2 are markedly downregulated in the ESCC setting, indicating a shift in their regulatory functions within tumors. The decreased expression of LHPP suggests a reduction in its tumor-suppressive effects, potentially facilitating ESCC pathogenesis. The diminished expression of HMOX1 could indicate a compromise in antioxidative defenses, possibly heightening oxidative stress—a condition that could be leveraged by proliferating ESCC cells. Moreover, the lowered expression of BCL2, typically involved in inhibiting cell death, hints at a subtle susceptibility of ESCC cells to apoptotic triggers, an aspect critical to understanding the survival strategies of ESCC cells.
In conclusion, these dynamic gene changes gene expression provide deep insights into the cellular transformations occurring during ESCC development. This s research sheds light on the intricate molecular drivers of these processes and opens avenues for further investigation and potential therapeutic interventions in ESCC.
STAT3 was pinpointed as a central regulator in the advancement of ESCCTo further explore the regulatory mechanisms underlying ESCC pathogenesis, we utilized a list of differentially expressed genes (DEGs) list derived from the ESCC disease database (https://tcga-xena-hub.s3.us-east-1.amazonaws.com/download/TCGA.ESCA.sampleMap%2FHiSeq.gz) and performed a comprehensive integrative analysis with our scRNA-seq dataset (Fig. 4A, B). Our analysis identified 14 consistently upregulated genes (STAT3, VEGF, MMP9, SPP1, HIF1A, TGFB1, EZH2, EGFR, PIK3CA, KRAS, HER2, MMP3, CDH2, CTNNB1) and 8 genes that were downregulated. Notably, the significant downregulation of LHPP, which encodes a phosphatase involved in the dephosphorylation of phospholysine and phosphohistidine residues, stood out. Previous research has linked its downregulation with tumor advancement and adverse outcomes. (Guo et al. 2022; Hindupur et al. 2018; Zhu et al. 2023) Further correlation analysis between LHPP and transcription factors of the differentially expressed genes showed a notable negative association with STAT3 and a positive correlation with PPARA (Fig. 4C, D).
Fig. 4
STAT3 was identified as a central regulator in ESCC progression. A Volcano plot showing the DEGs distribution in bulk RNA-sequencing data. B Venn plot showing the overlap between scRNA-seq DEGs and bulk RNA-seq data. C Scatter plot displays the correlation between LHPP and STAT3 expression across normal (orange dots) and ESCC (green dots) tissue samples. D Scatter plot displays the correlation between LHPP and PPARA expression across normal (orange dots) and ESCC (green dots) tissue samples. E The relative mRNA levels of the overlap DEGs were detected in 21 paired ESCC patient tissues
To validate our findings, we recruited a cohort of 21 cancer patients for histological confirmation, revealing significant mRNA upregulation of STAT3, IL6, MMP9, MMP3, TGFB1, VEGF and HIF1A, coupled with downregulation of LHPP and PCK1, PTEN and BLC2 (Fig. 4E). However, alterations in EZH2 and PPARA expression were not statistically significant. These findings suggest a potential transcriptional inhibitory role of STAT3 on LHPP expression.
STAT3 plays a negative regulatory role in LHPP expression in ESCCTo delve deeper into the regulatory roles of STAT3 in ESCC, we undertook transcription factor prediction using these differentially expressed genes. Our findings highlighted STAT3, SOX2, FOXO3, and PPARA as the most significant transcription factors, possess the largest number of target genes, underscoring their critical roles in gene regulation within ESCC (Fig. 5A). Coupled with the prediction analysis of the STAT3 transcription factor binding motif, we identified a binding site for STAT3 in the promoter region of LHPP (Fig. 5B).
Fig. 5
STAT3 Plays a Negative Regulatory Role in LHPP Expression in ESCC cells. A Dot plot showing the upregulated (red) and downregulated TFs, with size indicating the target gene number. B Representation predicted binding motif of STAT3. C Western blot analysis displaying the overexpression levels of STAT3 proteins in EC9706. D Luciferase reporter assay results showing the effect of STAT3 on the LHPP promoter activity in EC9706. E Luciferase reporter assays illustrating the lack of suppressive activity in promoter Mutation compared to the wild-type promoter in the presence of STAT3. F ChIP-qPCR analysis showing STAT3 binding to the PAFR and LHPP promoter in KYSE-150 cells
In the ESCC cell line EC9706, we employed Luciferase reporter assays to validate the binding of STAT3 to the LHPP promoter region. Our result confirmed the activation of the LHPP promoter in the presence of overexpressed STAT3, indicating a functional binding of STAT3 to this specific promoter region (Fig. 5C, D). These findings in the progression of ESCC disease indicate that STAT3 negatively regulates LHPP expression by binding to its promoter region, suggesting a significant transcriptional inhibitory role that contributes to the pathophysiology of the disease. To further dissect the mechanism underlying STAT3's influence on LHPP promoter activity, we introduced a point mutation at the putative STAT3 binding site within the promoter region (Fig. 5E). This mutation altered the consensus sequence, hypothesized to be critical for STAT3's interaction. The luciferase activity measurements post-mutation, shown in Fig. 5E, revealed a significant reduction to near-baseline levels, underscoring the essential role of this binding site for STAT3-mediated transcriptional regulation. Based on predictions from binding motifs and validations through luciferase reporter assays and site-directed mutagenesis, we identified a potential positive STAT3 binding site at Site2 (− 1786 to − 1991 bp) and a potential negative site at Site1 (− 546 to − 780 bp) in the promoter region of LHPP. Additionally, we used previously published ChIP-on-chip data from three ESCC cell lines, which revealed STAT3 binding sites on the PAFR gene, serving as an additional experimental control (Zhao et al. 2023). ChIP-qPCR results in KYSE-150 cells confirmed the enrichment of STAT3 at the LHPP promoter's Site2 and on the PAFR gene, aligning with our findings from luciferase assays. Notably, no enrichment was observed at the negative control site1, further validating the specificity of STAT3 binding in our study. This consistency between ChIP-qPCR and luciferase data reinforces the reliability of our conclusions regarding the regulatory interactions of STAT3.
Collectively, our findings highlight the profound capability of single-cell genomics to demystify the complex molecular landscape of ESCC. The identification of pivotal transcription factors such as STAT3, and their associated expression patterns, not only deepens our comprehension of the disease's pathophysiology but also directs us towards more precise and potentially effective therapeutic interventions. Importantly, the discernment of STAT3's regulatory role suggests its critical influence in the progression of ESCC, offering a promising target for future treatment strategies.
留言 (0)