
記住我
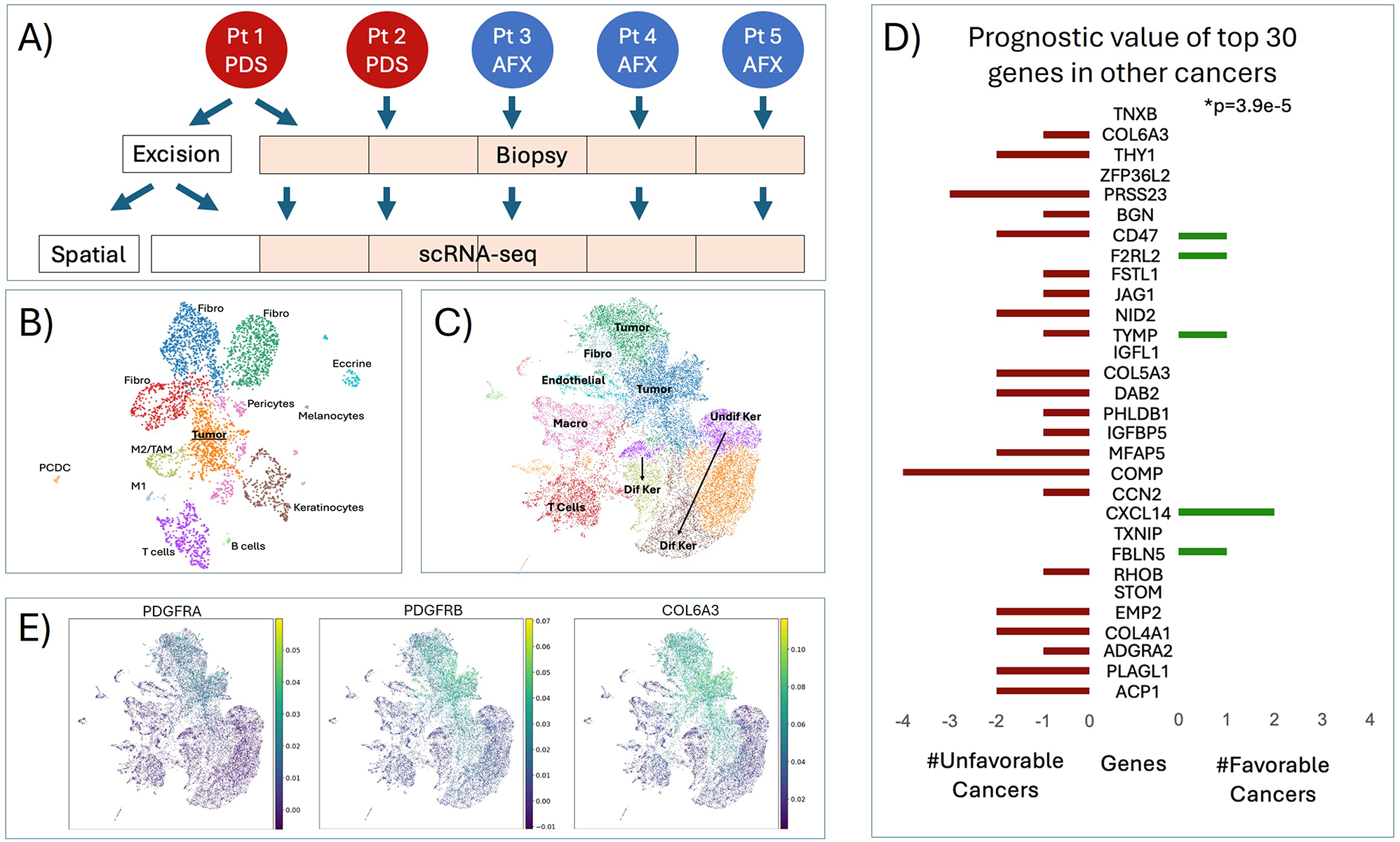

The CCF for each gene was calculated for 38 samples (19 primary (prima group), 19 metastatic (meta group)). The CCF value denotes the proportion of cells that contain the corresponding mutation. For example, if all the tumour cells in a specific tumour tissue carry a certain somatic mutation, then the CCF of that mutation is 1. In the Pyclone result file, the ‘cellular_prevalence’ value denotes the proportion of malignant cells with the mutation in the sample, which is also referred to as the CCF in the literature. The CCF_diff value denotes the difference in the mean CCF value for a specific gene between the meta group and the prima group. The CCF_PValue was obtained via a t test and denotes the significance of the difference in the CCF value between the prima and meta groups for a specific gene.
Exome capture analysisFor this analysis, the Genome Analysis Toolkit (GATK; version 4.2.6.1) was used for the detection of single-nucleotide polymorphisms (SNPs) and insertions/deletions (InDels). The MarkDuplicates tool [19] was utilized to identify duplicates and build an index. Base quality recalibration was performed via the BaseRecalibrator and ApplyBQSR tools, relying on known databases such as dbsnp_146.hg38.vcf.gz and Mills_and_1000G_gold_standard.indels.hg38.vcf.gz. Mutect2 was used for SNP and InDel calling. Copy number variations (CNVs) were called via CNVkit (version 0.9.8). Mutsig was employed to predict driver genes.
Whole-transcriptome sequencingRNA isolation and library preparationTotal RNA was extracted with the TRIzol (Thermo Scientific, USA) reagent according to the manufacturer’s protocol. RNA purity and quantity were evaluated with a NanoDrop 2000 spectrophotometer (Thermo Scientific, USA). RNA integrity was assessed with an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). A total of 1 µg of RNA per sample was used as the starting material for RNA sample preparation. Ribosomal RNA was removed with a Ribo-off™ rRNA Depletion Kit (Human/Mouse/Rat) (Vazyme, China). The sequencing libraries were subsequently generated with various index labels with the NEBNext® Ultra™ Directional RNA Library Prep Kit for Illumina (NEB, USA) following the manufacturer’s recommendations.
Sequencing of RNAs and identification of novel lncRNAsThe libraries were sequenced on the Illumina NovaSeq 6000 platform, and 150 bp paired-end reads were generated. Raw data (raw reads) in fastq format were first processed via fastp (version 0.20.0) software with default parameters in paired-end mode. The clean data were obtained for downstream analyses by removing reads containing adapter sequences, reads containing poly-N sequences and low-quality reads from the raw data. The clean reads were mapped to the human genome via HISAT2 with the parameters -p %s --fr --dta-cufflinks -S. After the reads were assembled via StringTie (version 1.3.3.b) with the parameters -p 8 --rf, novel transcripts were identified by comparing the known annotated genes with the reference genome via the Cuffcompare function with default parameters. To calculate the protein-coding ability of the novel transcripts, CPC, CNC, Pfam and PLEK software were used. The overlapping transcripts that were predicted to be noncoding transcripts by these four software programs and had a transcript length of > 200 nucleotides were defined as novel lncRNAs.
Differential RNA expression analysisThe clean reads were mapped to the human mRNA and lncRNA reference sets via Bowtie2 (version 2.3.1) with the parameters -k30 -t -p 20. The FPKM value and read count of each transcript were obtained via eXpress (version 1.5.1) with the parameters --no-update-check --rf-stranded. Differential expression analysis was performed via the DESeq (2012) R package. The functions estimateSizeFactors and nbinomTest were used to normalize the data and calculate the differential p values, respectively. A p value < 0.05 and a fold change > 2 or < 0.5 were set as the threshold criteria for significant differential expression. Hierarchical cluster analysis of the DEGs was performed to explore gene expression patterns. Gene Ontology (GO) term enrichment and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses of the DEGs were performed with R based on the hypergeometric distribution.
MeRIP-seqCell collectionA sufficient quantity of tissue or cells, typically approximately 5 to 20 × 10^6 cells or more than 100 mg of tissue, respectively, was collected for each MeRIP experiment.
RNA extraction and quality inspectionTotal RNA from each sample was isolated and purified with TRIzol (Invitrogen, CA, USA). A NanoDrop ND-1000 spectrophotometer (NanoDrop, Wilmington, DE, USA) was used for quality control analysis of total RNA quantity and purity. The integrity of the RNA was then assessed with a 2100 Bioanalyzer system (Agilent, CA, USA) and confirmed through agarose gel electrophoresis. The downstream experimental requirements included a concentration of > 50 ng/µl, an RNA integrity number (RIN) of > 7.0, an optical density (OD) 260/280 of > 1.8, and a total RNA quantity of > 50 µg.
IP and library constructionFirst, polyadenylated (PolyA) mRNA was captured from the sample by two rounds of purification with oligo(dT) magnetic beads (Dynabeads Oligo (dT), Catalogue No. 25-61005, Thermo Fisher, USA). The captured mRNA was fragmented under high-temperature conditions with a magnesium ion fragmentation reagent kit (NEBNext® Magnesium RNA Fragmentation Module, Catalogue No. E6150S, USA) at 86 °C for 7 min. The fragmented RNA was combined with beads from an immunomagnetic bead Dynabeads Antibody Coupling Kit (Thermo Fisher, CA, USA) and an anti-m6A polyclonal rabbit antibody (Synaptic System, Cat. No. 202003), an anti-m5C antibody (Diagenode, 5-methylcytosine (5-mC) Antibody-clone 33D3), an anti-m1A antibody (MBL, Anti-1-methyladenosine (m1A) mAb, D345-3) and an anti-m7G antibody (MBL, anti-7-methylguanosine (m7G) mAb, RN017M) for IP in premixed IP buffer containing 50 mM Tris-HCl, 750 mM NaCl, and 0.5% Igepal CA-630. The immunoprecipitated RNA was then reverse transcribed into cDNA with Invitrogen SuperScript™ II Reverse Transcriptase (Catalogue No. 1896649, CA, USA). E. coli DNA polymerase I (NEB, Catalogue No. m0209, USA) and RNase H (NEB, Catalogue No. m0297, USA) were used for double-strand (DS) synthesis to convert these DNA‒RNA hybrid double strands into double-stranded DNA. dUTP Solution (Thermo Fisher, Catalogue No. R0133, CA, USA) was added to the double-stranded DNA for end blunting, and a single A nucleotide was then added to both ends for ligation with a T-nucleotide-containing adapter. The fragments were then size selected and purified with magnetic beads before digestion of the double-stranded DNA with the UDG enzyme (NEB, Catalogue No. m0280, MA, US). Subsequent PCR amplification (denaturation at 95 °C for 3 min; 8 cycles of denaturation for 15 s at 98 °C, annealing for 15 s at 60 °C, and extension for 30 s at 72 °C; and final extension at 72 °C for 5 min) was carried out to generate a library with fragments of 300 bp ± 50 bp.
Sequencing and analysisA quality check of the constructed library was conducted. Sequencing was performed on the Illumina NovaSeq 6000 platform after the library passed quality inspection. Upon receiving the raw RIP-seq data, we employed fastp software to trim adapters and filter out contaminants to obtain clean data. Next, we applied HISAT2 software for genome alignment. Following deduplication, the data were used in exomePeak (https://bioconductor.org/packages/exomePeak) for peak calling and differential analysis. After the peaks were identified, ChIPseeker software was used for peak annotation, and MEME SUITE was utilized for motif analysis. StringTie was used to reconstruct and quantify the transcripts and for selection and enrichment analysis of differentially expressed transcripts.
RIP-seqSample collectionA sufficient amount of tissue or number of cultured cells, i.e., approximately 5 − 20 × 10^6 cells or more than 100 mg of tissue, was collected for each RIP experiment.
Cell lysisThe final cell pellet was resuspended in an equal volume of polysome lysis buffer composed of 100 mM KCl, 5 mM MgCl2, 10 mM HEPES (pH 7.2), 0.5% NP40, and 1 mM dithiothreitol (DTT) and RNase and protease inhibitors. The suspension was incubated on ice for 5 min. Cellular debris was then pelleted by centrifugation at 15,000 × g for 15 min at 4 °C. After centrifugation, 40 µl of the supernatant was collected for RNA extraction as an input sample, while the remaining supernatant was set aside for immunoprecipitation.
Immunoprecipitation(1) Antibody coating of protein A/G beads: First, 100 µl of protein A/G magnetic beads was washed with NT2 buffer (50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1 mM MgCl2 and 0.05% NP-40) three times. (2) Bead incubation: The magnetic beads were resuspended in 850 µl of the immunoprecipitation reaction solution, which contained 200 units of an RNase inhibitor, 400 µM ribonucleoside vanadyl complexes (RVC), 10 µl of 100 mM DTT, 30 µl of 0.5 mM ethylenediaminetetraacetic acid (EDTA) and 800 µl of cool NT2 buffer. The cleared cell lysates were placed in the immunoprecipitation reaction solution and incubated at 4 °C for 4 h on a tube rotator. (3) Bead washing: Subsequently, the beads were washed five times with 1 ml of ice-cold NT2 buffer.
SequencingTRIzol was added to the input and IP samples to isolate total RNA. RNA quality control was conducted with a Qubit fluorometer. Libraries for sequencing were prepared with the SMART-Seq v4 Ultra Low Input RNA Kit (Takara Clontech Kit, Cat# 63488). Finally, the libraries were sequenced on the NovaSeq 6000 platform (Illumina).
Construction of RJH-metastasis 1.0RJH-Metastasis 1.0 integrates a total of 43 selectable features across four major categories: the RNA level (25 features), DNA level (5 features), protein level (11 features), and literature evidence level (2 features). Each major category includes information from multiple different datasets.
Sample collectionAt the RNA level, transcriptome sequencing data and clinical information for 461 patients from the TCGA-COAD cohort were analysed. A total of 4 patients with CRC and 4 patients with CRLM were enrolled in three independent cohorts at Ruijin Hospital, Shanghai Jiao Tong University School of Medicine, between January 2018 and January 2020. Ethical approval for the study was obtained from Ruijin Hospital, Shanghai Jiao Tong University School of Medicine. Transcriptome sequencing, m1A meRIP-seq, m5A meRIP-seq, m5C meRIP-seq and m7G meRIP-seq were performed on the 8 samples from these patient cohorts. All the data were uploaded to the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO) public database (GSE271081). Additionally, the weight of genes highly expressed in non-small cell lung cancer was reduced on the basis of data derived from the GSE222334 dataset of NCBI GEO.
At the DNA level, two whole-exome capture sequencing datasets were obtained from TCGA-COAD and SRP041725. The CCF for each gene was calculated for 38 samples (19 primary (prima group), 19 metastatic (meta group)).
At the protein level, data were sourced from the TCGA Colon Cancer Proteome (https://pdc.cancer.gov/pdc/study/PDC000111), which contains data for 64 samples (58 M0 samples and 6 M1 samples).
At the literature evidence level, publications with publication dates from 1994 to 2020 and listed in three cancer metastasis literature databases were included. From the Human Cancer Metastasis Database (HCMDB; http://hcmdb.i-sanger.com/index), a total of 7337 cancer metastasis-related publications and 771 CRC metastasis-related publications were obtained. From CMGene (a literature-based database and knowledge resource for cancer metastasis genes, https://bioinfo-minzhao.org/cmgene/index.html), a total of 5596 publications were obtained. From the Tumor Metastasis Mechanism-associated Gene Database (TMMGdb; http://hmg.asia.edu.tw/TMMGdb/), a total of 15,323 cancer metastasis-related publications were obtained.
Signature constructionAt the RNA level, meRIP-seq analysis was used to detect differences in methylation modifications, including m1A, m6A, m7G, and m5A, of specific genes between metastatic samples and primary samples (threshold: fold change ≥ 2.0, P value ≤ 0.00001). Three features (hit, regulation, and fold change) of genes with methylation modifications were considered. In particular, genes with and without m1A modification differences were recorded as “m1A_hit = 1” and “m1A_hit = 0”, respectively. Genes with m1A modification differences and upregulated expression were recorded as “m1A_Regulation = 1”, and genes with m1A modification differences and downregulated expression were recorded as “m1A_Regulation = -1”. The fold change in the m1A modification difference was recorded as m1A_Foldchange. The features (hit, regulation, and fold change) of m6A, m7G, and m5A were recorded accordingly. The number of RNA modifications in a specific gene was recorded as the full_hit value. Differences in RNA expression between metastatic samples and primary samples were detected via RNA-seq. The upregulated genes in the metastatic samples were recorded as “regulation = 1”, and the downregulated genes in the metastatic samples were recorded as “regulation = -1”. The fold change in differential expression was recorded as the logFC value. The Pvalue based on DESeq analysis, and the corrected false discovery rate (FDR) were recorded. An RNA correlation network was constructed via correlation analysis (absolute Spearman correlation coefficient > 0.8, P < 0.05). Descriptive network metrics of each gene in the RNA correlation network were obtained via the “R igraph” package (https://igraph.org/r/doc/) and were selected as features, as follows: Degree, closeness_centrality, betweenness_centrality, eigenvector_centrality, page_rank_score, and clustering_coefficient. A total of 433 dynamic network biomarkers (DNBs) were identified on the basis of the TCGA-COAD cohort. Genes identified as DNBs were recorded as “DNB = 1”, and all other genes were recorded as “DNB = 0”. The average FPKM expression values of genes in 100 HCC samples were recorded as liver_exp values.
At the DNA level, the CCFs of mutated genes in metastatic samples and primary samples were first obtained. CCF_p_ttes and CCF_p_wilcoxon were determined via the t test and the Wilcoxon test. CCF_diff was calculated as the mean CCF value in the metastatic samples subtracted from the mean CCF value in the primary samples. CCF_abs_diff was the absolute value of CCF_diff. The Drivergene parameter was defined on the basis of three databases, namely, the SRP041725 dataset, TCGA-COAD dataset and Network of Cancer Genes website (http://ncg.kcl.ac.uk/index.php), and “drivergene = 1” indicated that at least two data sources identified the specific gene as a driver gene.
At the protein level, the differential expression profile of proteins was obtained via proteomic analysis on the basis of a dataset of CRC tissues (metastatic vs. primary). The average expression values of proteins in the metastasis group and primary group were recorded as protein_m1. mean and protein_m0.mean, respectively. protein_Diff was the average difference in the expression of a specific protein between the 2 groups, and protein_abs_Diff was the absolute value of protein_Diff. protein_pvalue was the p value determined via a t test. A PPI network was constructed on the basis of the differentially expressed proteins (https://cn.string-db.org). Descriptive network metrics of each protein in the PPI network were obtained via the “R igraph” package and were selected as features, as follows: protein_Degree, protein_closeness_centrality, protein_betweenness_centrality, protein_eigenvector_centrality, protein_page_rank_score, and protein_clustering_coefficient.
For the literature evidence level, two features based on the publications were selected. The number of publications in which the specific gene was reported was recorded as lit_meta. The number of CRC-related publications in which the specific gene was reported was recorded as lit_meta_colon.
Multiobjective recommendation: Pareto fronts were computed on the basis of the implementation approach in the rPref R package [20]. During multiobjective optimization, we looked for solutions that occupied an optimal surface (front), which represented the best trade-offs between the considered objectives. This optimal surface was labelled Pareto level 1. The RJH-Metastasis 1.0 interface returned genes that occupied Pareto level 1. The recommendation for CRLM in this study was obtained by adjusting 34 parameters (4 parameters: low and 30 parameters: high; Supplementary Table 2) and ignoring 9 parameters.
Sample collectionA total of 180 patients with CRC and 25 patients with CRLM identified between January 2018 and January 2020 were enrolled in three independent cohorts at Ruijin Hospital, Shanghai Jiao Tong University School of Medicine. Ethical approval for the study was obtained from Ruijin Hospital, Shanghai Jiao Tong University School of Medicine. Cohort 1 consisted of 155 CRC patients with available tissue microarray and immunohistochemical analysis data. Cohort 2 was composed of 25 CRC patients with available tissue microarray and immunohistochemical analysis data. Cohort 3 consisted of 25 CRLM patients who received anti-PD-1 immunotherapy (PD-1, 200 mg, every three weeks).
All diagnoses of CRC were based on histopathological evaluation and were in accordance with the World Health Organization (WHO) criteria. Tumour stage was determined via the Union for International Cancer Control (UICC) Tumor, Node, Metastasis (TNM) Classification System. Tumour recurrence was diagnosed on the basis of CT scans, histological confirmation, and an elevated CEA level.
Patients who underwent palliative surgery only, had prior interventions (e.g., transhepatic artery embolization or radiotherapy), or were diagnosed with other primary malignancies or inflammatory diseases during the follow-up were excluded from the study.
The single-cell transcriptome and single-cell spatial transcriptome data were obtained from the GSE225857 dataset. Single-cell transcriptome analysis was performed on sequencing data from paired primary colon cancer and liver metastasis samples from 5 patients (s0107, s0115, s0813, s0920 and s1231). Four primary CRC samples (C1-C4) and two liver metastasis samples (L1 and L2) were selected for spatial transcriptome sequencing.
RT-PCR analysisIn accordance with the TRIzol method [21], total RNA was extracted from SW480 cells and METTL1-knockdown cell lines, and the RNA was reverse transcribed into cDNA. Real-time fluorescence quantitative PCR was performed with a SYBR Green kit, and GAPDH was used as the internal reference. The sequences of the primers used for PCR were as follows: GNB1F (AGTGACCCTGTTTTGTGGCA), GNB1R (ACATCAGAGAGGCTGCCCTA), GNB1NF (GCTGTTTGACCTTCGTGCTG), GNB1NR (AAGTCGTCGTACCCAGCAAG), PHF1F (ATGGAGCACTCACCAGCTTC), PHF1R (GCTCCTCCACTTTCCCCTTC), DVL2F (CGTACTGGGTCAGTCCTGTC), DVL2R (ATCACCTTCGTCTCCCCAAC), GAPDHF (GATTTGGTCGTATTGGGCGC), GAPDHR (TTCCCGTTCTCAGCCTTGAC), METTL1F (TGGGACATCTAGGCACCTCA), METTL1R (CTGGTTTGGGAGGTCACTGC), PHF1NF (TGAGCAAGCCCCTCCTCTAT), PHF1NR (CAGGACAAGATGGGCCACAT), DVL2NF (AATGTGTCCAGCAGCCATGA), and DVL2NR (TCGCTGGTCATGAGGGTAGA).
ImmunohistochemistryThe tumour regions were stained with an anti-GNB1 (rabbit polyclonal, Abcam, Cat# ab137635), anti-METTL1 (rabbit monoclonal, Abcam, Cat# ab271063), or anti-Ki67 (rabbit polyclonal, Abcam, Cat# ab15580) antibody. First, the slides were baked at 60 °C for 1–2 h. The slides were then dewaxed by hydrophilic treatment in the following sequence: incubation in xylene-1 for 10 min, xylene-2 for 10 min, 100% anhydrous ethanol for 5 min, 95% ethanol for 5 min, and 75% ethanol for 5 min. Then, 5% hydrogen peroxide was added, and the slides were incubated for 15 min. Next, 1X antigen retrieval solution was prepared and added, and the samples were heated in a microwave oven at high heat for 10 min. Blocking solution was added to block nonspecific antigens, and the samples were incubated for 2 h. The primary antibody was added, and the samples were incubated at 4 °C overnight. The next day, the secondary antibody was added, and the samples were incubated at room temperature for 2 h. The slides were then stained with chromogenic reagent, the staining time and degree were monitored by microscopy, and at the appropriate time, the slides were immediately rinsed to stop the reaction. Haematoxylin staining of nuclei was terminated when the nuclei developed a marked blue colour, as evaluated by microscopy. Then, the samples were dehydrated and dried overnight. An appropriate amount of neutral resin was added to the sealing film, which was then allowed to dry for 1 day. All evaluations were performed by the same pathologist blinded to the patient characteristics.
Western blot analysisProteins were separated via 10% SDS‒PAGE and transferred to polyvinylidene difluoride membranes, and the membranes were then washed and blocked [22]. The membranes were incubated with anti-GNB1 (rabbit polyclonal, Abcam, Cat# ab137635), anti-METTL1 (rabbit monoclonal, Abcam, Cat# ab271063), anti-DVL2 (rabbit polyclonal, Proteintech, Cat No: 12037-1-AP), anti-PHF1 (Rabbit polyclonal, Proteintech, Cat No: 15663-1-AP), and anti-GAPDH (mouse monoclonal, Proteintech, Cat No: 60004-1-I) primary antibodies, followed by the corresponding secondary antibodies.
Cell proliferation and migration assaysCell proliferation was evaluated by a CCK-8 assay. Cells were seeded in 96-well plates (1500 cells/well) in 100 µl of medium, 10 µl of CCK-8 solution (Dojindo) was added, and the plates were incubated for 2 h. The number of viable cells was determined via the absorbance method (wavelength of 450 nm).
Cell proliferation was also evaluated by a colony formation assay. Cells (2000 cells/well) were plated in a 6-well plate. After 2 weeks of culture, the cells were fixed with paraformaldehyde and stained with crystal violet. Photographs were taken, and data were collected.
Transwell assays were used to evaluate cell migration. A 24-well plate containing membranes with 8-µm-diameter pores (Minipore) was used. A total of 700 µl of Dulbecco’s modified Eagle’s medium (DMEM; containing 10% FBS) was placed in the lower compartment, and 1 × 10^5 cells in 200 µl of DMEM (containing 1% FBS) were placed in the upper compartment. After 48 h, the cells remaining in the upper compartment were removed by wiping, and the cells that migrated to the lower surface of the membrane were fixed with 4% paraformaldehyde and counted in 5 microscopic fields of view (200× magnification).
Assessment of tumour growth and liver metastasis in vivoSW480, SW480-GNB1, SW620, and SW620-sh-GNB1 cells were injected into mice (1 × 106 cells, suspended in 200 µl of serum-free DMEM, right flank region). The animals were monitored twice weekly and were euthanized 35 days after cell implantation. Bioluminescence imaging was performed with an IVIS Lumina K Series III instrument, and image brightness values were normalized via the live images. Following sacrifice, the tumours were excised, the weight of each tumour was determined, and the lungs were harvested for bioluminescence imaging.
Single-cell transcriptome analysisThe Seurat package’s FindVariableGenes function (FastExpMean as mean.function, FastLogVMR as dispersion.function) was used to choose the top 2000 highly variable genes (HVGs). The expression profiles of these HVGs were then subjected to principal component analysis (PCA), with the results displayed in a two-dimensional space via UMAP, a tool for nonlinear dimensionality reduction.
For the identification of marker genes, the FindAllMarkers function in the Seurat package (with presto as test.use) was utilized. In this process, genes that were upregulated in each cell type compared with all other cell types – i.e., potential marker genes – were identified. Visualization of these identified marker genes was accomplished through the VlnPlot and FeaturePlot functions.
Cell type identification was performed via the SingleR package (version 1.4.1), wherein the expression profiles of the cells in question were matched against a common reference dataset. The cells to be identified were assigned to the cell type with the highest correlation in the reference dataset, thereby minimizing subjective influence. This identification approach involved the computation of the Spearman correlation coefficient between each cell in the sample and each annotated cell in the reference dataset, with the cell type showing the maximum correlation designated the final cell identity.
The Seurat package’s FindMarkers function (with presto as test.use) was used to choose DEGs for enrichment analysis. Genes with a fold change greater than 1.5 and a p value less than 0.05 were considered significantly differentially expressed.
Spatial single-cell transcriptome analysisQuality control and data preprocessing for gene quantification were executed via the Seurat package (version 4.3.0). The sctransform function in the package was used for data normalization and detection of high-variance features. These processed data were then stored in the SCT matrix.
The methodology for dimensionality reduction and clustering analysis utilized the FindVariableGenes function, a feature of the Seurat package, to single out the top 3000 HVGs. The expression profiles of these HVGs were subsequently subjected to (PCA. The PCA results were then plotted in two-dimensional space via the UMAP algorithm, an effective tool for nonlinear dimensionality reduction.
RCTD (version 1.1.0), a robust cell type deconvolution method, was used for spatial annotation of cell types. RCTD uses cell type profiles sourced from scRNA-seq data to decompose combinations of cell types while simultaneously correcting for inter-sequencing technology variability. During the RCTD operation, the creat.RCTD function was used with its standard parameters, guaranteeing that each cell type contained at least one cell and ensuring that every spot carried at least one unique molecular identifier (UMI). The run.RCTD function was used with doublet_mode set to FALSE, enabling the computation of the cell type composition within each spot.
Multiplex immunofluorescence (mIF) stainingWe conducted mIF staining using antibodies specific for GNB1 (rabbit polyclonal, Abcam, Cat# ab137635), CD20 (rabbit monoclonal, Abcam, Cat# ab64088), CLEC2C (CD69) (rabbit monoclonal, Abcam, Cat# ab307081), CD8 (rabbit monoclonal, Abcam, Cat# ab217344), KLRB1 (CD161) (rabbit monoclonal, Abcam, Cat# ab302564), and PD1 (rabbit monoclonal, Abcam, Cat# ab237728). Xylene was used to deparaffinize the tissue sections, which were then rehydrated with ethanol. Then, the tissue sections were subjected to antigen retrieval by boiling in Tris-EDTA buffer (pH 9.0) for 15 min. Endogenous peroxidase activity was blocked by incubation with 3% hydrogen peroxide at room temperature for 15 min. Nonspecific antigens were blocked by incubation with goat serum solution for 30 min. The sections were then incubated with primary antibodies overnight at 4 °C and with horseradish peroxidase (HRP)-conjugated secondary antibodies at room temperature for 30 min. The sections were subsequently incubated with Opal tyramide signal amplification (TSA) fluorochromes (Opal Colour Manual IHC Kit, Perkin Elmer, NEL811001KT) at 37 °C for 20 min. Between each run, the antibody (Ab)-TSA complexes in the sections were removed via microwave heating, and the sections were blocked with goat serum solution. In the final run, 4’,6-diamidino-2-phenylindole dihydrochloride (DAPI) was added for visualization of nuclei, and the sections were mounted with glycerin.
Statistical analysisGraphPad Prism 9.0 (San Diego, CA, USA) was employed for plotting and statistical analysis. The t-test and Mann–Whitney test were applied for normally and non‐normally distributed data, respectively. The OS represented the time from diagnosis to the last follow‐up or death. For continuous variables, student’s t‐test was used to analyse the differences between two groups, One-way ANOVA and Tukey’s post hoc test were used to analyse the difference among multiple groups. The data are displayed as the mean ± SEM from three independent experiments. A P value < 0.05 was considered to indicate statistical significance unless otherwise noted.
Additional methods can be found in the Supplementary Information.
留言 (0)