
記住我
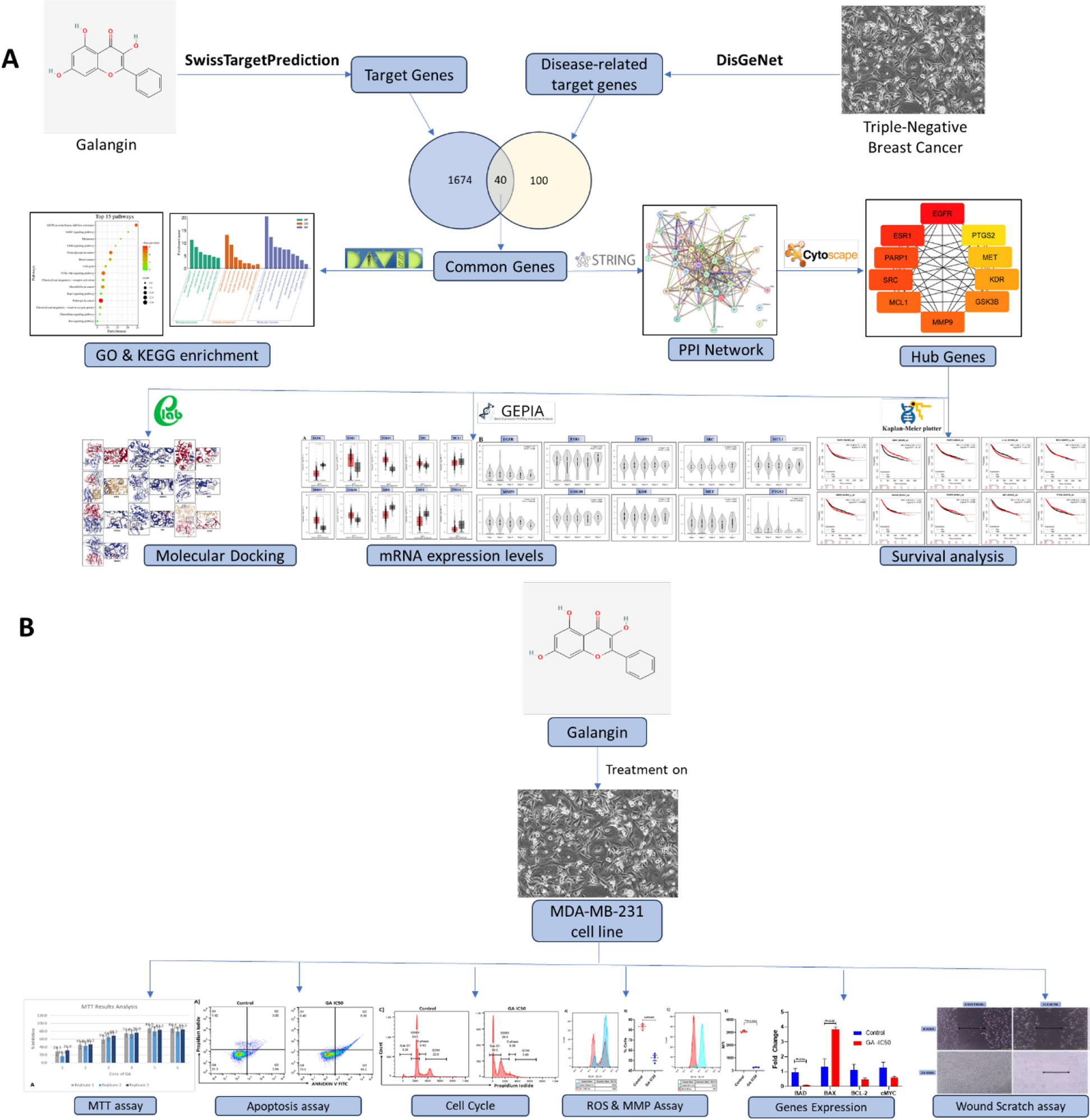








Among the 598 samples analyzed in the TCGA-LUAD project, 13 patients had no clinical and survival data. Therefore, RNA-sequencing expression profiles and other information from 59 normal and 526 LUAD samples were included in this study. The LUAD samples were randomly split into two groups: a training cohort with 421 samples and a testing cohort with 105 samples. Table S1 details the clinical characteristics of samples in the training, testing, and entire cohorts, indicating no significant differences among them (P > 0.05).
Screening of DE-IRGsAccording to the adjusted P-value < 0.01 and |log2 (fold change)|> 2, a total of 696 DEGs were identified between normal and LUAD samples of the TCGA-LUAD project for further analysis (Fig. 2A). After integrating 1565 IRGs, we obtained 91 DE-IRGs (Fig. 2B), of which 20 DE-IRGs were up-regulated, and 71 DE-IRGs were downregulated (Table S2). DE-IRGs expression profile of normal and tumor samples is shown in Fig. 2C.
Fig. 2
Identification of DE-IRGs between LUAD samples and normal samples. A Volcano plot of DEGs based on TCGA-LUAD project. B Venn diagram for the intersections between LUAD DEGs and IRGs. C The heatmap of DE-IRGs expression between the normal and tumor samples
Functional enrichment analysisTo better understand the underlying mechanisms and predict the prognosis of LUAD, we further investigated the functions and pathways affected by these 91 DE-IRGs. GO analysis indicated that the most significantly (adjusted P-value < 0.05) enriched terms for biological process, molecular function, and cellular component were “regulation of chemotaxis,” “G protein-coupled peptide receptor activity,” and “external side of plasma membrane,” respectively. The most ten highly enriched terms for the three ontologies are represented in Fig. 3A and Table 1. To identify possible signaling pathways associated with DE-IRGs, we conducted an analysis of KEGG with data from the TCGA cohort (Fig. 3B and Table 2). The results of ToppFun enrichment are also summarized in Table S3.
Fig. 3
Functional enrichment analyses of DE-IRGs. A Most significant enriched Gene ontology (GO) categories for the validated DE-IRGs. B The enriched pathways of the DE-IRGs
Table 1 The list of 10 most significant enriched GO categories for DE-IRGs. (Adjusted P-value < 0.05)Table 2 The list of most significantly enriched pathways for DE-IRGs. (Adj P-value < 0.05)Construction of prognostic prediction model based on risk scoreA univariate Cox regression analysis was performed to recognize potentially predictive genes among DE-IRGs, and 10 DE-IRGs were found to have significant relations to OS (P < 0.05) in the training cohort of LUAD patients (Table 3). Next, the candidate genes underwent LASSO Cox regression analysis to eliminate genes with high correlations and minimize overfitting. Totally, 9 of the 10 DE-IRGs were screened (Fig. 4). The heatmap of these 9 DE- IRGs between two risk groups in the entire cohort is depicted in Fig. 5. We utilized these 9 DE-IRGs to construct the prognosis predictive model by multivariate Cox regression analysis (Table 4) and calculated the risk score as follows:
Fig. 4
Construction of prognostic prediction signature. LASSO regression was performed to identify the minimum criteria
Fig. 5
The heatmap of 9 DE- IRGs between two risk groups in the total cohort
Table 4 Coefficients and multivariable cox model results of 9 IRGs in risk signature$$\mathrm\;\mathrm\:=\:(\mathrm5\;\exp.\:\times\:0.081428)\:+\:(\mathrm\;\exp.\:\times\:0.050255)\:+\:(\mathrm S100\mathrm P\;\exp.\:\times\:0.047464)\:+\:(\mathrm3\;\exp.\:\times\:\:-\:0.008143)\:+\:(\mathrm1\;\exp.\:\times\:\:-\:0.040922)\:+\:(\mathrm\;\exp.\:\times\:\:-\:0.026052)\:+\:(\mathrm1\;\exp.\:\times\:\:-\:0.082896)\:+\:(\mathrm4\;\exp.\:\times\:0.159715)\:+\:(\mathrm4.1\;\exp.\:\times\:\:-\:0.083087)$$
Validation of the prognostic prediction modelTo validate the immune-related gene signature, the entire testing cohorts were used as internal validation to verify its predictive capability. All LUAD patients within the three cohorts were stratified into low- and high-risk groups using the median risk score value derived from the training group. As the next step, we investigated how well the prognostic model could distinguish survival in patients’ risk groups. The analysis of Kaplan–Meier curves indicated a significant difference in OS among the two predicted groups of all cohorts, and high-risk patients had a poor outcome (Fig. 6A–C). The time-dependent ROC curves were performed to validate the accuracy of the model, and the 5-year AUC values gained 0.684, 0.717, and 0.689 in the training, testing, and entire cohorts, respectively (Fig. 6D–F), suggesting that it may be feasible to predict the survival of LUAD patients using this presented model. Additionally, the findings indicate that patients with higher risk scores are more prone to worse survival outcomes (Fig. 6G–L). Overall, these results showed satisfactory predictive performance of the IRGs signature in TCGA-LUAD data. We also validated our model using an external independent validation dataset (the GSE68465 dataset). Of note, the Kaplan–Meier curves analysis demonstrated a more satisfactory outcome for low-risk group patients. The results of Kaplan–Meier curves, as well as 1-, 3- and 5-year AUC, were represented in Fig. 7, further highlighting that the risk signature performed satisfactorily as a predictor of external data.
Fig. 6
Validation of the immune-related signature in the TCGA cohort. A–C The Kaplan–Meier curve analysis of the high- and low-risk groups in the training, testing, and total cohorts. D–F ROC curve analysis of the prognostic prediction model in the training, testing, and entire cohorts. G–L The distribution of risk scores and survival status in the training, testing, and total cohorts
Fig. 7
Validation of the immune-related signature in the GSE68465 cohort. A The Kaplan–Meier curve analysis of the high- and low-risk groups in GEO cohort. B ROC curve analysis of the prognostic prediction model in GEO cohort
Evaluation of the prognostic prediction modelAssociation between the model and clinicopathological featuresInterestingly, the results of the Wilcoxon rank sum test indicate a statistically significant association between the risk score and clinicopathological features of patients. Specifically, the 9-IRG risk score demonstrated a notably elevated correlation with advanced clinical T stage (P = 0.0482) and N stage (P < 0.0001) (Fig. 8C, D). Accordingly, the prognostic value of the model may partially be due to its association with clinicopathological characteristics.
Fig. 8
The relationships between the immune-related risk signature and A age; B clinical stage; C T stage; D N stage; E M stage
Independent prognostic role of the modelUnivariable Cox and multivariable Cox were used to analyze the effects of patients’ clinicopathological factors on the predictive value of the risk score as an independent parameter. Although the advance clinical stage, TNM stage, and high score of risk were factors that made OS unfavorable, the most significant association was seen between the OS and the risk score in the multivariable Cox analysis (HR = 2.5700, P = 2.36e-06) (Table 5), indicating the independent prognostic value of IRG signatures—regardless of age, disease stage, and TNM stages—in LUAD patients.
Table 5 The univariate and multivariate cox regression analysis to evaluate the independent prognostic valueAssociation between the risk score and tumor-infiltrating immune cellsOverall, the high-risk group demonstrated lower frequencies of immune cells. As represented in Fig. 9, not only DC and MQ but also T lymphocytes reduced in the tumor microenvironment of high-risk patients, suggesting that impaired antigen presentation to T cells may at least partly contribute to poor prognosis. Accordingly, NK cells—as the most important innate immune cells against cancer cells—were low in these patients. We also applied the Wilcoxon rank sum test on the results of XCELL and quanTIseq to investigate the association between the risk groups and tumor-infiltrating immune cells, depicted in Fig. S1.
Fig. 9
The correlation between risk score and tumor-infiltrating immune cells, which were analyzed by different quantification methods of immune infiltration estimations including CIBERSORT, quanTIseq, TIMER, XCell
Association between the risk score and mutation profileIn the examination of LUAD patient mutation statuses, we have identified the top ten most significantly mutated genes for both high- and low-risk groups. These findings are visually represented in Fig. 10A and B. In the following analysis, we computed the TMB for each sample. Our findings revealed a considerably higher TMB in the high-risk patients (P = 3.148e-06) (Fig. 10C); however, we did not observe any relationship between TMB and OS (P = 0.81) (Fig. 10D). The results of this section will be further elaborated in the Discussion.
Fig. 10
Tumor mutational burden (TMB) status among risk groups. A Mutation profile of the low-risk group. B Mutation profile of the high-risk group. C A correlation analysis between TMB and risk score. D The Kaplan–Meier curve analysis of high- and low-TMB groups
Association between the risk score and response to ICIIt has been confirmed that IPS could serve as predictive markers in melanoma patients undergoing treatment with PD-1 and CTLA-4 blockers [24]. Given this, it was tempting to investigate whether there is a relationship between our immune model and IPS. As represented in Fig. 11, the IPS scores exhibited a significant increase within the low-risk 9-IRG group, indicating a more pronounced immunogenic phenotype in this particular low-risk cohort. Furthermore, patients with a low risk had elevated levels of CTLA-4 (P = 1.913e-08), PD-1 (P = 8.118e-05), PDL-1 (P = 0.0243), and PDL-2 (P = 0.007663) expression, suggesting that ICI could be a promising treatment option for low-risk LUAD patients.
Fig. 11
The association between risk groups and response to immune checkpoint inhibitors (ICI). A The gene expression of CTLA-4, PD-1, and PD-L1 in the high-risk and low-risk groups. B The association between IPS and the immune-related risk signature in LUAD patients
留言 (0)