Explanation of the choice of comparators
The is an opinion, which is based on very few researches, that prolapse in the anterior, apical, and posterior compartment reconstruction of I and II level by DeLancey should be accompanied by reconstruction of the perineal body (which is in the III level) in order to prevent the recurrence of pelvic organ prolapse in the future [9, 10]. However, there is not enough evidence to prove that opinion. The aim of this trial is to evaluate the efficiency and safety of simultaneous perineoplasty on the clinical and anatomical efficacy of mesh-augmented sacrospinal fixation in advanced pelvic organ prolapse repair. Based on previous studies, it was difficult to estimate and comprehend whether colpoperinoplasty actually reduces the risk of prolapse recurrence.
We chose the following lines of comparison:
1.
Mesh-augmented sacrospinal fixation with posterior colporrhaphy (Hybrid pelvic floor reconstruction) and perineoplasty
2.
Mesh-augmented sacrospinal fixation with posterior colporrhaphy (Hybrid pelvic floor reconstruction)
Intervention description
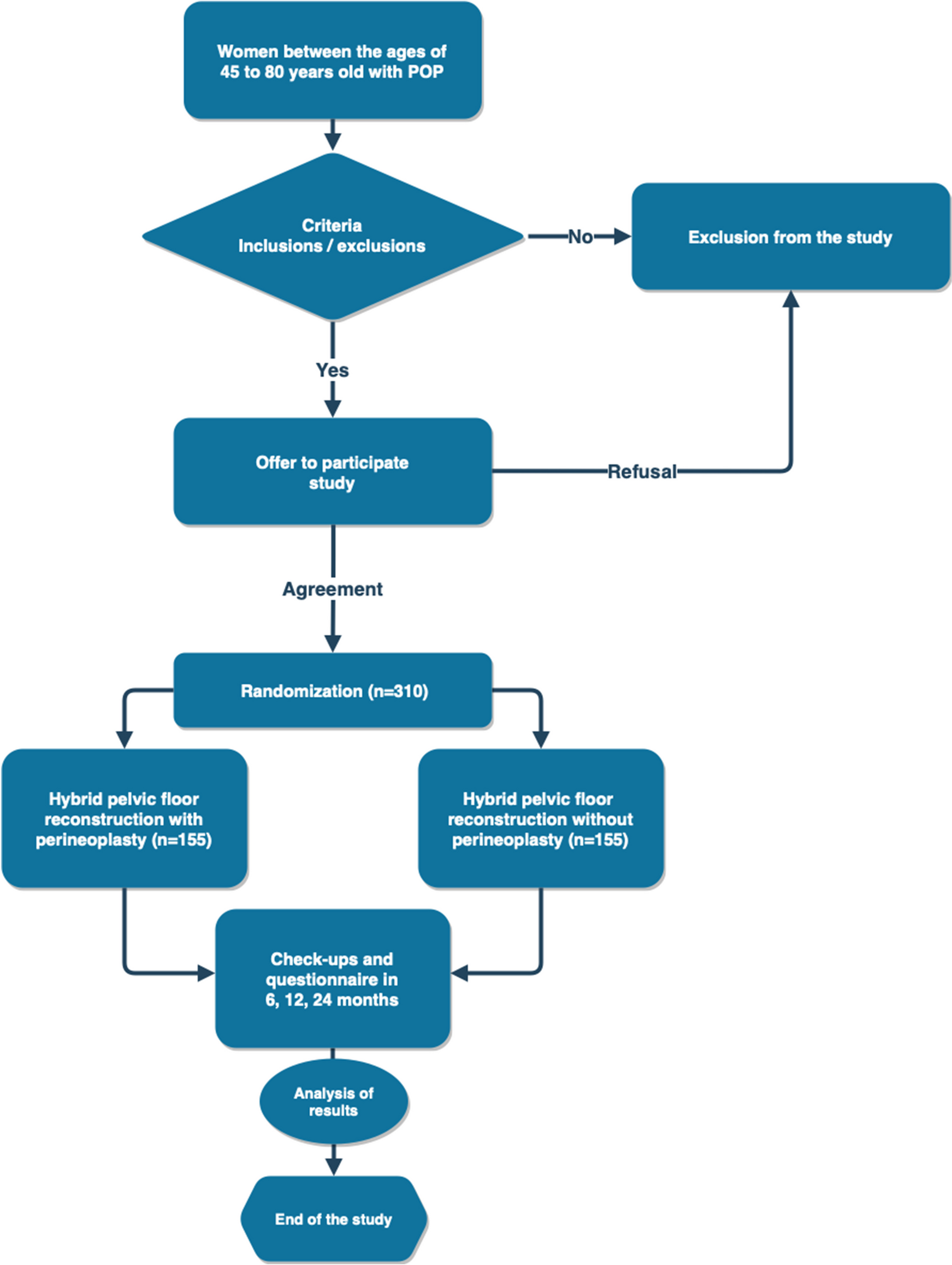
Patients who met the inclusion/exclusion criteria will be randomized into 2 groups:
1st group—patients who will undergo pelvic floor reconstruction (unilateral sacrospinal fixation using a mesh implant, anterior subfascial colporrhaphy, posterior subfascial colporrhaphy).
Description of the method: in the lithotomy position, after preparation of the surgical field and installing a urethral catheter, hydropreparation of the anterior wall of the vagina is performed with saline. A median incision is made in the anterior wall of the vagina, dissection of the paravaginal tissues in the direction of the left or right sacrospinous ligament. The endoprosthesis “Urosling 1” © (Lintex, St. Petersburg) is passed through the sacrospinous ligament using the Urofix PL instrument using the “inside-out” technique, fixed to the fibrous ring of the cervix with interrupted sutures using Ftorex suture (USP 2). Hemostasis and the integrity of the rectum and bladder are monitored. To correct a defect in the pubocervical fascia, anterior subfascial colporrhaphy is performed with a PGA corset suture (USP 2). Then a suture on the wound of the anterior wall of the vagina is applied using PGA (USP 0). After hydropreparation of the posterior vaginal wall with saline, a median incision is made in the posterior vaginal wall, subfascial dissection of the paravaginal tissues, then posterior subfascial colporrhaphy with a PGA corset suture (USP 2). Then a suture on the wound of the posterior wall of the vagina is applied using PGA (USP 0) [11]. A distinctive feature of surgery in this group is the absence of reconstruction of the perineal body and paravaginal tissues below the level of the hymenal ring.
2nd group—patients who will undergo pelvic floor reconstruction (unilateral sacrospinal fixation using a mesh implant, anterior subfascial colporrhaphy, posterior extended subfascial colpoperineoplasty).
Description of the method: reconstruction of the anterior and apical defects of the pelvic floor support is identical to the surgical technique in the first group. The difference in the reconstruction technique of the posterior compartment lies in the fact that the median incision of the posterior wall of the vagina, dissection of the paravaginal tissues is made to the level of the posterior commissure of the vagina with the transition of the dissection to the perineal body. In order to correct a defect in the recto-vaginal fascia and the tendon center of the perineal body, posterior subfascial colporrhaphy is performed with a corset suture to the level of the posterior commissure of the vagina with the transition of a continuous suture to the perineal body. After that simple sutures are applied to the perineal body with PGA (USP 2), suture on the posterior wall and 3rd row of sutures on the perineal body are applied using PGA (USP 0). For the skin, we use simple sutures PGA (USP 3/0) [11].
For both groups, the chosen method of anesthesia is endotracheal anesthesia. The urinary catheter and tampon with Levomekol ointment, installed after the operation, will be removed the next morning. Urinary control will be carried out 2, 4, 6, and 12 h after the removal of the urethral catheter). The criterion for the recovery of urination will be the volume of residual urine less than 50 ml, according to ultrasound. If urination is not restored, the patient will be discharged with a urinary catheter under the supervision of a urologist at the place of residence. To exclude formed hematomas in the area of operation, an ultrasound examination of the pelvic organs will be performed the next day.
Criteria for discontinuing or modifying allocated interventions
Drug dose change in response to the participant’s request.
Strategies to improve adherence to interventions
Drug tablet return.
Relevant concomitant care permitted or prohibited during the trial
All patients will be given antibacterial prophylaxis before surgery, prevention of thromboembolic complications in the form of the mandatory use of compression stockings, and the appointment of low molecular weight heparins. To relieve pain after surgery, nonsteroidal anti-inflammatory drugs will be prescribed, and if they are ineffective, opioid analgesics will be prescribed.
At the outpatient stage of rehabilitation, patients will be allowed to sit, but a strict restriction of physical activity and abstinence from sexual activity for 2 months is recommended.
Provisions for post-trial care
Not applicable; no post-trial care is used in this trial.
Outcomes Primary clinical endpoint
1.
Objective cure rate
The patient will be considered cured if, during postoperative follow-up, there is no recurrence of POP requiring repeated surgical treatment, an objective criterion is assessed according to the POP-Q classification (0–1 stage).
Secondary clinical endpoints
1.
Surgery satisfaction
Will be estimated with the “Global Impression of Improvement questionnaire” (PGI-I), validated in Russia. The patient marks the number that best describes her post-operative condition, compared with how it was before surgery. The score ranges from 1 (very much better) to 7 (very much worse).
2.
Influence of operation on sexual function.
Will be estimated with the Pelvic Organ Prolapse/Urinary Incontinence Sexual Questionnaire (PISQ-12), validated in Russia.
3.
Influence of operation on the quality of life.
Will be estimated with Pelvic Floor Disability Index (PFDI-20), validated in Russia.
4.
Complications
The presence of any complications, such as bleeding requiring blood transfusion, hematoma requiring drainage, organ perforation, nerve damage, accompanied by corresponding clinical manifestations, cicatricial deformity, shortening of the vagina, wound infection, urinary tract infection, chronic pelvic pain, extrusion of the implant into the vagina, urethral implant erosion, de novo dyspareunia, de novo urgency, bladder atony, and stress incontinence of urine de novo.
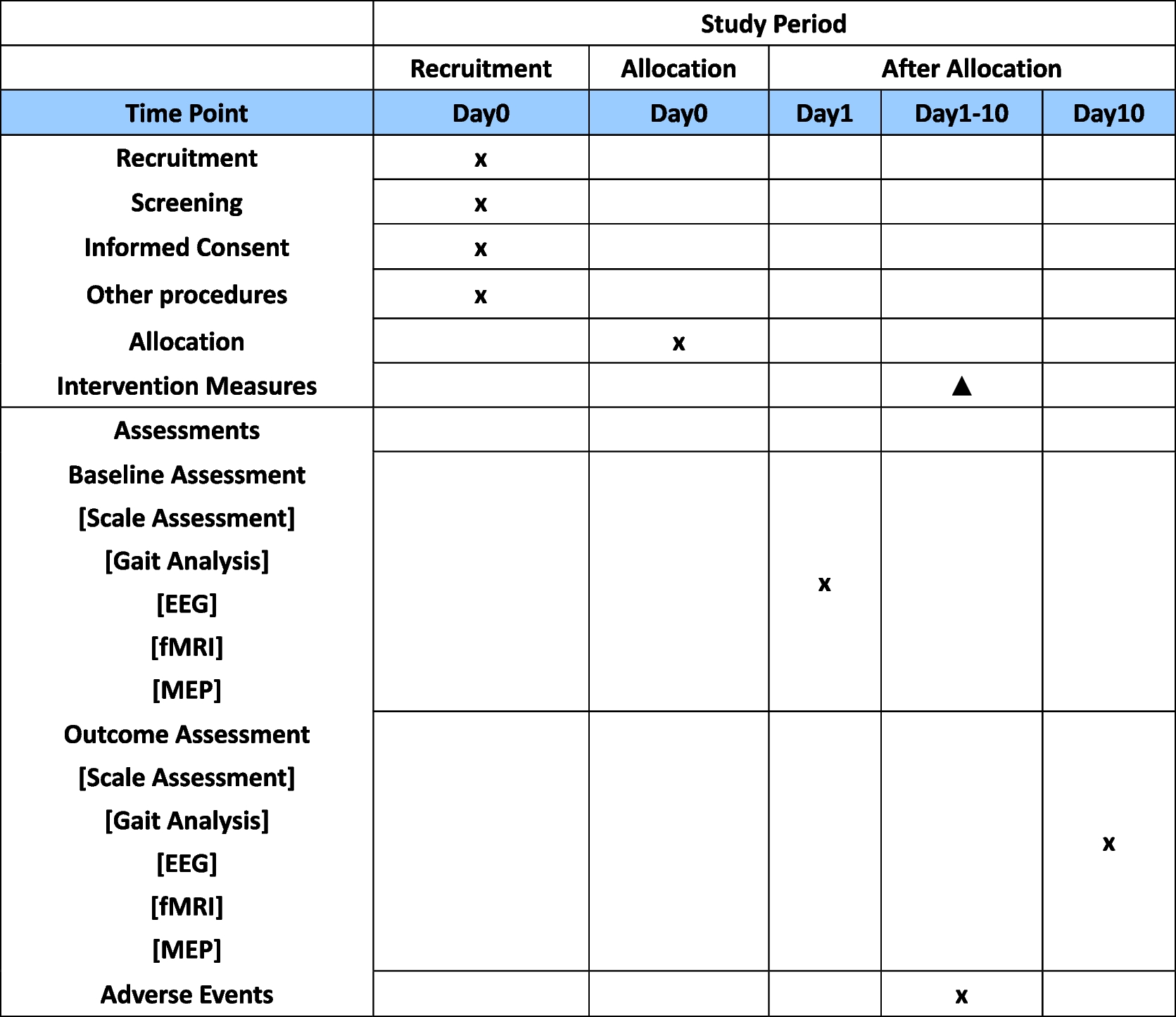
Factors, baseline values, and parameters that may affect the course of the postoperative period will also be assessed, such as duration of symptoms, medical history, body mass index, smoking, pre- or post-menopausal status, use of estrogen or hormone replacement therapy, repeat surgery for POP or stress urinary incontinence, and the use of a pessary (Table 1).
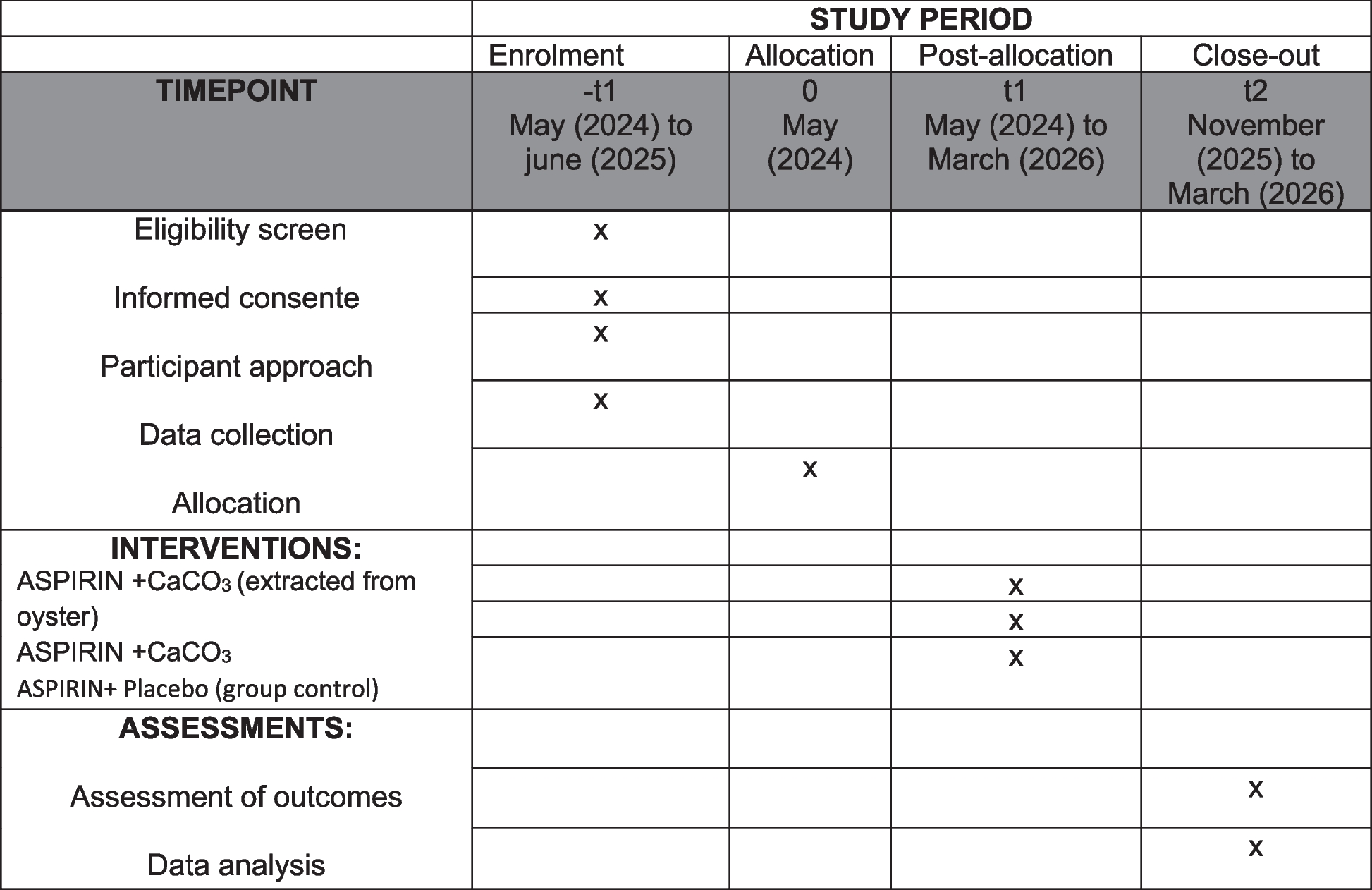
Participant timeline
Patients will be called to an appointment 6, 12, and 24 months after discharge, they will be examined and phoned for information about the long-term period.
Completion date: within 24 months after selection.
Sample size and
Taking into account the available data on the recurrence rate of unilateral sacrospinous fixation using this technology (7.4%) [11], as well as clinical observations on the recurrence rate of three-level reconstruction (1%), study power 80%, and significance level 5%, 282 patients are needed to confirm the expected difference in recurrence rate. To compensate for data loss, the estimated sample size is increased by 10%. As a result, the total sample size is 310 patients.
Recruitment
In SPBSU It is estimated that 2000 patients with POP are seen each year, while a smaller number become inactive due to relocation, change of health care provider, etc. Once identified in the center, patients potentially eligible for a specific study are contacted by the nurse coordinator who explains the study and ascertains the patient’s interest. If interested, the patient is seen by a urologist in the center and recommended for a trial if eligible.
Exclusion criteria
Patients who will no attend a follow-up visit within 2 months of the scheduled date and who will not answer the phone will be excluded from the study, as well as patients with a newly diagnosed cancer, decompensation of a chronic disease that may affect the study, as well as patients who die in the duration of observation.
Sequence generation
An Internet resource will be used for randomization https://www.sealedenvelope.com/simple-randomiser/v1/lists with the usage of the block randomization method with the size: 1 block–4 patients.
Participants will be randomly assigned to either control or experimental group with a 1:1 allocation as per a computer-generated randomization.
Concealment mechanism
We consciously chose not to make the trial “blind” because of the impossibility to hide operative methods from patients and medical personnel. However, the doctor who is in charge of collecting the data and examining patients 6, 12, and 24 months after the operation would not know in which group the patient was sorted into.
Implementation
All patients who give consent for participation and who fulfill the inclusion criteria will be randomized. Randomization will be requested by the staff member responsible for recruitment and clinical interviews from the coordinating center. After that, the patient will be randomized by the staff member using the internet resource https://www.sealedenvelope.com/simple-randomiser/v1/lists. Participants will be randomly assigned to either the control or experimental group with a 1:1 allocation as per a computer-generated randomization. The urologist in charge of postoperative interviews would not know in which group the patient was sorted into.
Who will be blinded
Not applicable; no blinding was used in this trial.
Procedure for unblinding if needed
The design is open-label with only outcome assessors being blinded so unblinding will not occur.
Plans for assessment and collection of outcomes
The initial data will contain demographic characteristics (age, sex, body mass index), in addition, patients at the prehospital stage will be asked to complete a survey using standard questionnaires validated in the Russian Federation: Pelvic Floor Disability Index (PFDI-20), Patient's Global Impressions of Improvement (PGI-I), Pelvic Organ Prolapse/Urinary Incontinence Sexual Questionnaire, IUGA-Revised (PISQ-12) [12, 13].
All communication including protocol modifications will be conducted by the corresponding author.
Plans to promote participant retention and complete follow-up
Once a patient is enrolled in this trial and randomized, the study site will make every reasonable effort to follow the patient for the entire study period. It is projected that the rate of loss-to-follow-up on an annual basis will be at most 10%. Study site staff are responsible for developing and implementing local standard operating procedures to achieve this level of follow-up.
Data management
All data will be collected by staff not involved in patient care. Base, procedural, and intraoperative data will be prospectively collected and reported in the form of a patient report. Everything will be stored electronically (database) using appropriate software. Original study forms will be entered and kept on file at SPBSU.
Confidentiality
Patients will be identified in the database using a unique code obtained after enrollment in the study. Patient reporting forms will only be in the form of initials and date of birth. Informed consent and contact details (for a 30-day follow-up telephone contact) will be kept separate from other records containing medical or other personal information.
Plans for collection, laboratory evaluation, and storage of biological specimens for genetic or molecular analysis in this trial/future use
Not applicable, no biological specimens are going to be used in this trial (Table 2).



留言 (0)