Cell lines
Human embryonic kidney cells (HEK293T), human keratinocytes (HaCaT), and HEK-Blue IL22 cells were cultured in high-glucose Dulbecco’s modified Eagle’s medium (DMEM) (BioSera, Cholet, France) supplemented by 10% fetal bovine serum (FBS) and streptomycin-penicillin solution (BioSera, Cholet, France), and incubated under 5% CO2 at 37 °C. Streptavidin-phycoerythrin (PE) was purchased from eBioscience, San Diego, CA, USA. Human anti-IL-22R1 APC-conjugated antibody (mouse IgG1 isotype) was procured from R&D Systems, Minneapolis, MN, USA, and mouse IgG1 APC-conjugated isotype control antibody MOPC-21 was obtained from Exbio Praha, a.s., Vestec, Czech Republic.
Assembly of ABD library
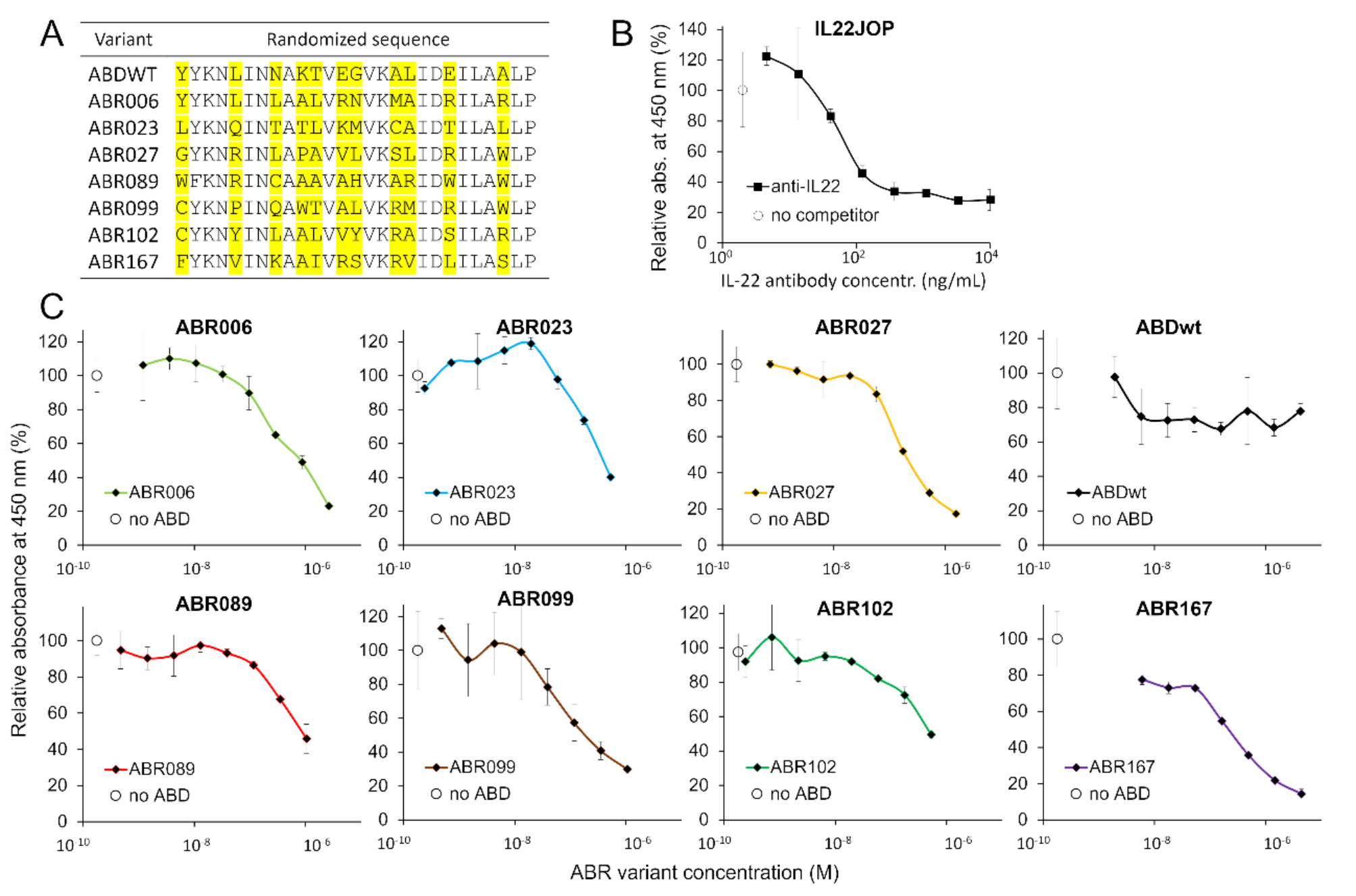
A highly complex combinatorial NNK library (where N means any nucleotide, K is G or T only) derived from a scaffold of albumin-binding domain (ABD) was assembled by multiple PCR steps as previously reported and characterized [38]. Briefly, the ABD library was assembled using two oligonucleotides ABDLIB-setB1c and ABDLIB-setB2c (Table S1) and connected with cDNA coding for the tolA helical linker. Amplified by PCR was purified on 1% agarose (QIAquick Gel Extraction Kit, Qiagen, Hilden, Germany) and used for in vitro transcription and translation using PURExpress® In Vitro Protein Synthesis Kit (NEB, Ipswich, MA, USA).
Ribosome display
A modified ribosome display selection procedure was used for the assembled ABD library targeting recombinant human IL-22R1 receptor [34, 36]. For the preselection step, 3% BSA in TBS buffer (50 mM Tris, 150 mM NaCl, DEPC-treated water, pH 7.5), and for the selection steps, IL-22R1 (rhIL-22 Rα1, R&D Systems, Minneapolis, MN, USA) diluted in coating buffer (Bicarbonate/carbonate coating buffer, pH 9.6), were coated in wells of NUNC-immune MaxiSorp plates (Nunc A/S, Roskilde, Sjælland, Denmark). Then, all the wells were blocked using Protein-Free (PBS) Blocking Buffer (Thermo Fisher Scientific, Waltham, MA, USA) and incubated for 3 h at room temperature (RT). Following, the preselection wells were washed thrice and incubated with a mixture of in vitro translated ABD library (mRNA-ribosome-protein complex) and ABDwt (25 µg/ml) in WBT buffer (50 mM Tris-base, 150 mM NaCl, 50 mM MgAc, 0.05% Tween-20, DEPC-treated water, acetic acid to pH 7.5) at 8 °C for next 1 h under shaking conditions. Later, the unbound library from preselection wells was transferred into selection wells, washed thrice, and incubated for 1 h at 8 °C under shaking conditions. Finally, the selection wells were washed thrice with WBT buffer while mRNA-ribosome-protein complexes bound to the coated IL-22R1 protein were eluted using 200 µl of EB (50 mM Tris-base, 150 mM NaCl, 50 mM EDTA, Diethyl pyrocarbonate (DEPC)-treated water, pH by acetic acid to pH 7.5). Next, the total mRNA was extracted and purified using mRNA extraction kit. A total of three rounds of preselection/selection were performed with increasing stringency as follows: 1st round – plate coating with 25 µg/ml IL-22R1, washed 5 times by WBT buffer containing 0.05% Tween 20, 2nd round – 25 µg/ml IL-22R1, washed 10 times and 3rd round – 10 µg/ml IL-22R1, washed 10 times by WBT containing 0.1% Tween 20. Also, ABDWT protein was added as a competitor to the library in 1st and 3rd rounds only. Harvested mRNA was reversely transcribed by GoScript™ Reverse Transcriptase (Promega, Madison, WI, USA) and resulted in a DNA library that was finally inserted into the pET28b vector to form a plasmid library of selected DNA variants.
Screening of IL-22R1-targeted ABD-derived variants
Escherichia coli BL21 (λDE3) BirA strain cells were transformed by the plasmid cDNA library. Individual bacterial colonies were picked up for overnight culturing in 2 ml LB broth at 37 °C with kanamycin (60 µg/ml) and chloramphenicol (30 µg/ml). Next day, 100 times diluted culture was further cultured, and protein production was induced with 1.5 mM isopropyl β-d-1-thiogalactopyranoside (IPTG) in the presence of 50 µM d-biotin (5 mM d-biotin solution in 10 mM Bicine buffer, pH 8.3) at 32 °C. After 4 h of incubation, the cells were harvested by centrifugation and obtained bacterial pellets were frozen. For the analysis, pellets were resuspended in PBS buffer and sonicated for 1 min using ultrasonic disruptor Misonix S3000 sonicator and centrifuged (18000×g) for 10 min at 4 °C. Cell supernatant was diluted 15,000 times in PBS/Tween-20/1%BSA (pH 7.4). For the identification of IL-22R1 specific binders, binding ELISA was used. Nunc MaxiSorp and PolySorp ELISA plates (Nunc A/S, Roskilde, Sjælland, Denmark) were coated with IL-22R1 (2.5 µg/ml). Then, MaxiSorp plates were blocked with Protein-Free (PBS) Blocking Buffer and PolySorp plates were blocked with 1% BSA in PBS-T (PBS, 0.05% Tween-20). Bacterial lysates containing in vivo biotinylated ABD protein variant were tested for binding to immobilized IL-22R1 using streptavidin-HRP conjugate (Pierce™ High Sensitivity Streptavidin-HRP, Thermo Fisher Scientific, Waltham, MA, USA). After 30 min incubation, TMB-Complete 2 (3,3’,5,5’-Tetramethylbenzidine, TestLine, Brno, Czech Republic) substrate was added and the reaction was stopped by 2 M sulfuric acid. The absorbance was measured at 450 nm using Epoch 2 microplate spectrophotometer (BioTek, Santa Clara, CA, USA).
Competition ELISA
ABD-derived variants were purified from bacterial lysates in 50 mM Tris, 300 mM NaCl, and pH 8.0 buffer using affinity chromatography (Ni-NTA agarose, Qiagen, Hilden, Germany). For the competition assay, MaxiSorp plates were coated with human IL-22R1 (1 µg/ml), blocked with Protein-Free (PBS) Blocking Buffer, and purified ABD-derived variants were serially diluted in PBS-T with 1% BSA containing IL-22 cytokine (recombinant human IL-22 protein, Fc Chimera, Abcam, Cambridge, United Kingdom) at the concentration 1 nM, and cytokine binding was detected by mouse IgG1 anti-human IgG (Fc) HRP (1: 5 000, mouse monoclonal IgG to Fc part of human IgG heavy chain conjugated with horseradish peroxidase, Exbio Praha, a.s. Vestec, Czech Republic).
Immunofluorescence staining of transfected HEK293T cells
Full-length human IL-22R1 cDNA coding for 574 amino acids protein containing a signal peptide (GenBank: BC029273.1) was cloned in pcDNA™6/myc-His A vector with added Kozak sequence. Before seeding the cells, 24-well plates (TPP, Trasadingen, Switzerland) were coated with 100 µl of 100 µg/ml poly-D-lysine (Gibco, Thermo Fisher Scientific, Waltham, MA, USA), incubated for 1 h at 37 °C, then washed 3 times with PBS and left to dry for 20 min at RT. Total 1.5 × 105 cells were seeded 48 h or 2.5 × 105 cells were seeded 24 h before the transfection. The complete growth medium was exchanged with DMEM without any supplements. Plasmid DNA and cationic polymer polyethylenimine (PEI) mixes were prepared in 50 µl of DMEM medium without supplements, mixed by inversion, incubated for 15 min at RT, and then applied dropwise into the wells. HEK293T cells were transiently transfected with PEI (PEI branched, MW 25,000; Sigma-Aldrich, St. Louis, MO, USA) in concentration 1 mg/ml, and in 4.5:1 PEI to plasmid DNA ratio, and 1 µg of DNA was used per each well. Transfected cells were incubated in 0.5 ml of DMEM medium without supplements for 18 h and then 0.5 ml of complete culture medium was added to each well. Two days after the transfection, particular protein variants at a concentration of 20 µg/ml were added to DMEM medium, and cells were incubated at 37 °C for 1 h and then 3 times washed with PBS. The primary staining mix contained Streptavidin–Alexa Fluor (AF) 568 conjugate (Thermo Fisher Scientific, Waltham, MA, USA) for detection of biotinylated ABR proteins and rabbit polyclonal anti-IL-22R1 antibody for the detection of IL-22R1. After 1 h incubation, cells were washed, and goat anti-rabbit IgG conjugated with AF488 (Abcam, Cambridge, United Kingdom) was added for next 1 h staining in the dark. Each well was washed 3 times with PBS and visualized using fluorescence microscopy with further image processing using ImageJ software.
Competition staining with ABR proteins in the presence of IL-22 competitor
Staining procedure was performed in the same manner as described in section “Immunofluorescence staining of transfected HEK293T cells” with the following modifications: before the staining, cells were treated with IL22 (Abcam, Cambridge, United Kingdom) for 1 h (cells were kept in cell incubator at 37 °C) which was followed by addition of ABR proteins into wells with previously treated and non-treated cells. Cells were again incubated for 1 h in cell incubator at 37 °C. After the incubation, cells were subjected to staining and visualized under microscope.
Binding kinetics measured with LigandTracer
HEK293T cells were transiently transfected with 1 mg/ml PEI (MW 25,000) and the PEI – DNA ratio was 4.5:1. Prior to 24 h before the transfection, 3 ml of cell suspensions (1 × 106 cells) was seeded on 100 mm cell dishes (NunclonTM, Sigma-Aldrich, St. Louis, MO, USA) in the designated area (marked as target area) and incubated overnight in a tilted position. Before seeding the cells, dishes were coated with 2.3 ml of 100 µg/ml poly-D-lysine, incubated for 1 h at 37 °C, then washed 3 times with PBS and left to dry for 20 min. The next day, the medium was exchanged into a medium without supplements and cells were transfected. Plasmid DNA and PEI mixes were prepared in 300 µl of DMEM medium without supplements, mixed by inversion and incubated for 15 min at RT. Transfected cells were incubated in 3 ml of DMEM medium without supplements in a tilted position for 18 h and then 6 ml of complete growth medium was added. Cells were incubated at the horizontal position for the next 24 h. For the measurement of ABR proteins kinetics, the LigandTracer Green Line (Ridgeview Instruments AB, Uppsala, Sweden) with Red - NIR (632–671 nm) detector was used. The fluorescence signal was detected using Streptavidin-APC conjugate (Thermo Fisher Scientific, Waltham, MA, USA). The evaluation of binding kinetics was done using TraceDrawer 1.7.1 software. Kinetic parameters (ka, kd, KD) were calculated using ‘One-to-one’ or ‘One-to-one depletion corrected’ evaluation methods.
Binding of ABR proteins to HaCaT cells
HaCaT cells were cultured in DMEM with 2 mM L-glutamine, 5.4 g/l glucose, 10% heat-inactivated fetal bovine serum, 100 U/ml Penicillin, 100 µg/ml Streptomycin at 37 °C in 5% CO2. After overnight incubation, 10 ng/ml TNFα (Abcam, Cambridge, United Kingdom) and 10 ng/ml IFNγ (Abcam, Cambridge, United Kingdom) were added for 24 h according to the previous study [39]. Biotinylated ABR variants were added in DMEM at a concentration 20 µg/ml (≈ 500 nM) for 1 h at 37 °C. Cells were washed 3 times with PBS. Rabbit anti-IL-22 polyclonal Ab (Abcam, Cambridge, United Kingdom) and rabbit anti-SARS-CoV-2 S1 RBD polyclonal antibody (RayBiotech, Peachtree Corners, GA, USA) as an isotype control were diluted 100 times in PBS with 1.5% BSA and added to cells and incubated for 1 h at RT. After 3 times washing with PBS, Streptavidin (AF568) (Invitrogen, Waltham, MA, USA) or goat anti-rabbit IgG H&L (AF488) pAb (Abcam, Cambridge, United Kingdom) were added to cells and incubated for 1 h at RT. Both reagents were diluted 200 times in PBS with 1.5% BSA. Finally, HaCaT cells were washed 5 times with PBS. Then, cell imaging was made with an Olympus CKX41 Inverted Phase Contrast Microscope (Olympus, Tokio, Japan). For the estimation of binding kinetics by LigandTracer, Streptavidin-APC conjugate was used for ABR proteins detection.
Flow cytometry
Streptavidin-phycoerythrin (PE) was obtained from eBioscience, San Diego, CA, USA. Human anti-IL-22R1 APC-conjugated antibody (mouse IgG1 isotype) was purchased from R&D Systems, Minneapolis, MN, USA and mouse IgG1 APC-conjugated isotype control antibody MOPC-21 was obtained from Exbio Praha s.r.o., Vestec, Czech Republic. Cultured HEK-Blue IL22 and HEK-293T cells were collected and washed in HEPES-buffered salt solution (HBSS buffer; 10 mM HEPES (pH 7.4), 140 mM NaCl, 5 mM KCl) supplemented with 2 mM CaCl2, 2 mM MgCl2, 1% (w/v) glucose, and 1% (v/v) FCS (cHBSS buffer). 2 × 105 cells/sample in cHBSS buffer were incubated with 1 µg/ml of biotin-labeled ligands (ABRs and ABD) for 30 min at 4 °C. After washing with cHBSS buffer, cells were incubated with PE-labeled streptavidin (diluted 1:400) for 30 min at 4 °C. Cells were washed, resuspended in cHBSS buffer, and analyzed by flow cytometry using a FACS LSR II instrument (BD Biosciences, San Jose, CA, USA) in the presence of 1 µg/ml of Hoechst 33258. Data was processed using the FlowJo software (BD Biosciences, Ashland, OR, USA) and appropriate gating was used to exclude debris, cell aggregates, and dead cells (Hoechst 33258-positive staining). Binding data are expressed as mean fluorescence intensity (MFI) values. For antibody binding, 2 × 105 cells/sample were incubated in cHBSS buffer with anti-IL-22R1 APC-conjugated antibody (diluted 1:100, final concentration 0.1 µg/ml) or IgG1 isotype control (diluted 1:100) for 30 min at 4 °C. After washing, cells were resuspended in cHBSS buffer, and analyzed by flow cytometry as described above.
HEK-Blue IL22 reporter cell inhibition assay
HEK-Blue IL22 Reporter Cell line (InvivoGen, San Diego, CA, USA) was cultured in DMEM with 2 mM L-glutamine, 5.4 g/l glucose, 10% heat-inactivated fetal bovine serum, 100 U/ml Penicillin, 100 µg/ml Streptomycin, 100 µg/ml Normocin, 10 µg/ml Blasticidin, 10 µg/ml Puromycin, 100 µg/ml Zeocin at 37 °C in 5% CO2. For the inhibition assay, HEK-Blue IL22 cells were seeded to a NUNC sterile 96-well plate (Nunc A/S, Roskilde, Sjælland, Denmark), 36,000 cells per well in a volume of 180 µl. HEK-Blue IL22 cells were grown on a surface treated with Poly-D-Lysine (Gibco, Thermo Fisher Scientific, Waltham, MA, USA) to ensure uniform attachment without cell clamping which was observed on an untreated surface. Inhibition assay was carried out in DMEM with 2 mM L-glutamine, 5.4 g/l glucose, 100 U/ml Penicillin, 100 µg/ml Streptomycin, 100 µg/ml at 37 °C in 5% CO2. Cells were incubated with human IL-22 (Abcam, Cambridge, United Kingdom) for 22 h in the presence of different concentrations of ABR variants. Neutralizing anti-IL-22 monoclonal antibody IL22JOP was used as a positive control, while ABDWT protein and irrelevant rat IgG2aκ anti-IL-6 monoclonal antibody (BioLegend, San Diego, CA, USA) were used as negative controls. ABR variants were serially diluted in sterile PBS by 5 times per step with the highest concentration being 120 nM and added to cells in 20 µl volume. After 22 h, 20 µl of cell supernatant was mixed with 180 µl of the Quanti-BlueTM Solution (InvivoGen, San Diego, CA, USA) to detect secreted alkaline phosphatase (SEAP) that was produced by HEK-Blue IL22 cells in response to human IL-22 stimulation. Cell supernatant was incubated with Quanti-Blue Solution for 1 h at 37 °C in the dark. Absorbance was measured at 620 nm with Epoch 2 microplate spectrophotometer. HEK-Blue IL22 reporter cells were incubated with 3 ng/ml (≈ 0.2 nM) of human IL-22 and varied concentration of ABR variants. Secreted embryonic alkaline phosphatase (SEAP) level for each ABR concentration, as detected by measuring absorbance at 620 nm, was compared to the SEAP level while human IL-22 was added in the absence of any other protein.
Molecular modeling
We modeled the structure of the ABD-derived variants using the MODELLER 9v14 software suite [40] using the structure of the wild type ABD (pdb id 1gjt [41]) as the template. The IL-22R1 structure was obtained from the crystal structure of the IL-22/IL-22R1 complex (pdb id 3dlq [9]). For protein-protein docking with flexible side chains, we utilized a local version of the ClusPro server [42, 43], using chain B from the 3dlq structure (residues missing in the template were modeled using the automodel function of MODELLER taking the best scoring model corresponding to IL-22R1 domains 1 and 2, residues 18 to 228, according to the UniProt [44] record Q8N6P7) as the receptor and the modeled ABR variants as ligands. The docking results were visualized with PyMOL version 2.6.0 (The PyMOL Molecular Graphics System, Schrödinger, LLC, New York, NY, USA).
Animals and experimental design
8–9 weeks old female C57BL/6 mice (AnLab, Prague, Czech Republic) weighing between 18 and 22 g (weight before treatment) were kept under standardized conditions at temperature between 21 and 22 °C, a 12:12-h light/dark cycle, ad libitum access to food and water. Mice were permitted to acclimatize for 1 week before starting the experiments. Mice were randomly divided into experimental groups and marked with ear tags.
Induction of acute colitis
Acute colitis was induced by giving drinking water containing 2.5% (w/v) DSS (MW approximately 40 kDa; TdB Labs, Uppsala, Sweden) for 4 days. At the end of the experiment, the animals were anesthetized, bled out, and euthanized by cervical dislocation. The length of the colon was measured between the caecum and proximal rectum. Serum was isolated from blood. Colon was dissected into pieces for quantitative real-time RT-PCR (qRT-PCR) and histochemistry. All experiment protocols were approved by Ethics Committee of the Faculty of Medicine and Dentistry (Palacky University Olomouc, Czech Republic), and the Ministry of Education, Youth and Sports, Czech Republic (MSMT-10947/2021-3).
Preventive treatment of mice by intraperitoneal application (i.p.) of binders
C57BL/6 mice were divided into three groups. Group 1 (naive) – consisting of 10 naive untreated mice. Group 2 (DSS + ABR167) – consisting of 19 mice treated by i.p. administration of 25 µg of ABR167 per mouse every 24 h starting 3 days before DSS-colitis induction and continuing for another 4 days together with DSS administration, and the Group 3 (DSS induction control) – consisting of 13 mice exposed to DSS without the therapy.
RNA isolation, reverse transcription, and qRT-PCR
Dissected colon tissues were stored in RNA later (Invitrogen, Waltham, MA, USA) at -80 °C. Pre-weighed colons were homogenized using Qiashredder (Qiagen, Hilden, Germany) and total RNA was extracted using RNeasy Plus Mini Kit (Qiagen, Hilden, Germany). Column DNase treatment (gDNA Eliminator spin column, Qiagen, Hilden, Germany) was used to eliminate potential DNA contamination. RNA purification was performed via lithium chloride method [45] to remove the traces of DSS. Extracted RNA was reversely transcribed into cDNA using the gb Elite Reverse Transcription Kit (Generi Biotech, Hradec Kralove, Czech Republic). qRT-PCR was performed in triplicates using gb SG PCR Master Mix (Generi Biotech, Hradec Kralove, Czech Republic) on LightCycler 480 System (Roche, Basel, Switzerland). All primers (6 µM) used in this study are listed in Table S2. Ct values were normalized to the reference gene, GAPDH, and the relative RNA expression was calculated by 2−ΔΔCt method [46].
Measurement of serum cytokine concentration
Concentration of cytokines IL-1β, IL-4, IL-6, IL-12p40, IL-18, IL-22, IL-23 and TNFα in serum was measured using fluorescent microbeads (LEGENDplex™ Custom Human Assay, Biolegend, San Diego, CA, USA) conjugated with antibody targeting particular cytokine according to manufacturer´s instructions. Briefly, serum and mixture of cytokine standards were applied into 96-well plate and diluted 1:1 in assay buffer. Mixture of conjugated beads was added into wells and incubated for 2 h at room temperature. Wells were washed and mixture of biotinylated detection antibodies targeting all selected cytokines was added. Plate was incubated for 90 min at RT. Then, streptavidin conjugated with phycoerythrin was added and plate was incubated for 30 min at RT. After washing, beads were analyzed using flow cytometer SONY SP6800 (Sony, Tokio, Japan).
Histochemistry
Formalin-fixed, paraffin embedded tissues were cut, stained with hematoxylin and eosin (H&E) (Merck, Darmstadt, Germany), classified by a professional pathologist, and verified by medical doctor without prior knowledge of clinical parameters using BX43 microscope equipped with CCD camera (Olympus, Tokio, Japan). The representative areas of the most intense mucosal alterations were selected in each section. These areas were scored for given parameters as summarized in the Table S5.
Statistical analysis
All statistical analyses were performed using GraphPad Prism 8 Software (GraphPad Software Inc., San Diego, CA, USA) or OriginLab version 2023b (OriginLab Corporation, Northampton, MA, USA). All data sets were verified for normal (Gaussian) distribution by Normality test. We performed Kruskal-Wallis one-way ANOVA followed by Dunn´s multiple comparisons test; *P < 0.05, **P < 0.01, ***P < 0.001.





留言 (0)