
記住我






To examine the expression patterns of Arid1a following brain injury, we firstly analyzed GSE129927 dataset in the Gene Expression Omnibus (GEO) database and found that Arid1a, Arid2 and Arid3a transcripts are increased following brain injury (Fig. S1a). However, the baseline expressions of Arid2 and Arid3a are considerably low (Fig. S1b), indicating that only Arid1a may play a crucial role after brain injury.
To test whether Arid1a is involved to brain injury, we used the already established TBI model in our lab [26] in which the cortex and hippocampus of mice were pierced with a No. 15 blade, resulting in an acute brain trauma (Fig. 1a-b). Quantitative PCR analysis indicated that Arid1a mRNA levels was significantly elevated in injured brain tissue at 3 days post-injury (dpi) (Fig. 1c), suggesting that Arid1a might be involved in the process of brain injury. To better understand the cell origin of Arid1a up-regulation post-TBI, we conducted RT-PCR analysis of mRNA levels of marker genes for cells within the lesion area and found that the mRNA expression of Aif1 (Iba1, a marker for microglia/macrophages) was the most significantly up-regulated, while NeuN (neuronal cell marker) and Olig2 (oligodendrocyte marker) mRNA levels were not altered in the injurer tissues at 3 dpi, compared to the sham group (Fig. 1d). Therefore, we postulated that the increased Arid1a expression observed after TBI was likely from Iba1+ microglia/macrophages. Macrophage infiltration is widely observed following TBI [29, 30]. To distinguish between microglia and infiltrating macrophages at lesion sites, we conducted IF staining for Tmem119 (a specific marker for microglia) and Iba1 at 3 dpi. We found that over 90% of Iba1+ cells around the injury area were Tmem119+/ Iba1+ cells, suggesting that the majority of Iba1+ cells accumulating in the lesion area after TBI are indeed microglia, rather than infiltrating macrophages (Fig. S2a-b).
Fig. 1
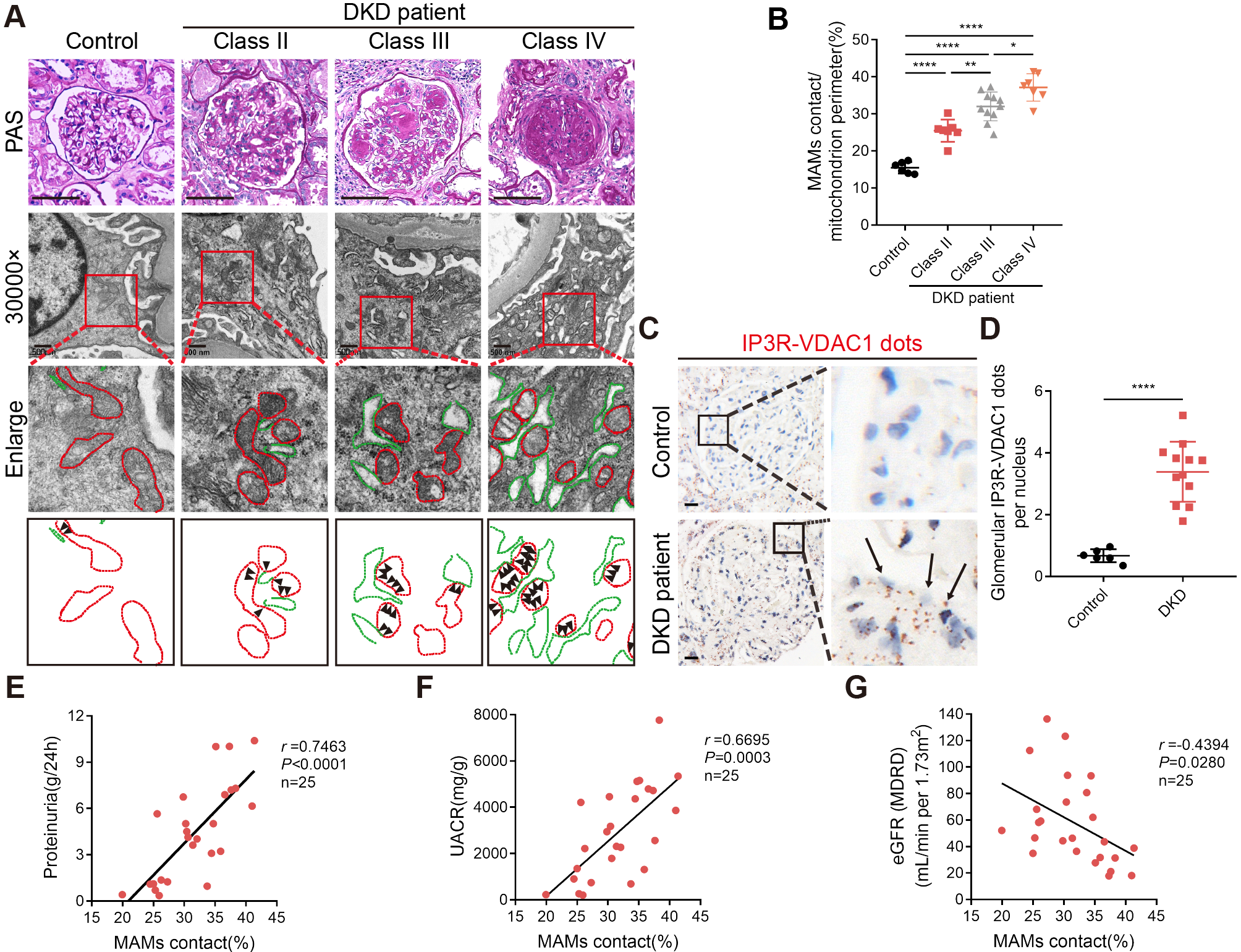
ARID1A expression is increased in microglia following brain injury in mice. a Schematic diagram of the mouse TBI model. b Photograph of a PBS-perfused mouse brain post-TBI. Scale bars: 5 mm. c qPCR analysis of Arid family genes in the brain tissues at 3 dpi. n = 4. d Relative mRNA levels of marker genes for cells within the lesion area at 3 dpi. n = 4. e-f Representative images (e) and quantification of ARID1A immunostaining (f) in microglia at 3 dpi. n = 4. Scale bars: 50 μm and 10 μm. Data are presented as mean ± SEM; ***P < 0.001, **P < 0.01 by Student’s t-test
To validate the cellular composition at lesion sites, we further performed co-immunofluorescence (IF) staining on brain sections at 3 dpi utilizing anti-ARID1A and anti-Iba1, anti-GFAP (a marker for astrocyte) or anti-NeuN antibodies. Notably, no significant accumulation of astrocytes or neurons within the lesion area was observed, and also no significant alterations in ARID1A expression were observed in astrocytes or in neurons (Fig. S3a-d). However, the quantification results clearly showed ARID1A expression was dramatically increased in Iba1+ cells surrounding the injury core (Fig. 1e-f), suggesting the up-regulation of ARID1A following TBI was primarily attributed to microglia.
Selective deletion of Arid1a in microglia exacerbates severity of glial scar formationThe cells in the CNS orchestrates a complex array of responses after TBI [31, 32]. A pathological feature of TBI is the formation of the glial scar, resulting from the rapid recruitment of microglia, in tandem with astrocytes, to the perilesional region where cellular necrosis is most prominent [33]. To explore whether ARID1A plays a functional role in glial scar formation, we used an Arid1a microglial cKO mice by crossing Arid1afl/fl mice with transgenic Cx3cr1-Cre mice [25] (Fig. S4a), and performed a comprehensive histological analysis at different time points after TBI. As we expected, ARID1A protein expression was nearly undetectable in the primary microglia isolated from Arid1a cKO pups (Fig. S4c-d). According to the experimental design in Fig. 2a, brain tissues were collected at specified time following the injury 3, 7, 21 and 42 dpi after undergoing either TBI or sham procedure. Our IF staining demonstrated that the numbers of both microglia and astrocytes were not significantly increased at lesion sites in either WT and Arid1a cKO mice at 3 dpi, but both microglia and astrocytes dramatically increased at lesion sites in the Arid1a cKO group at 7, 21 and 42 dpi (Fig. 2b).
Fig. 2
Selective deletion of Arid1a in microglia exacerbates the formation of glial scar after TBI. a Illustration of timeline for brains collection after TBI. b Representative images of IF staining of Iba1 (red), GFAP (green) and DAPI (blue) in brain slices at 3, 7, 21 and 42 dpi, respectively. Mouse brains were sectioned at 40-μm thickness into coronal sections. n = 5 animals per group. Scale bars: left panel, 1,000 μm; right panel, 100 μm. c, d Quantification of Iba1+ area (c) or GFAP+ area (d) at 3, 7, 21 and 42 dpi. n = 5 animals per group. Data are presented as mean ± SEM; ns, no significance; **P < 0.01, *P < 0.05 by Student’s t-test
Next, we carefully mapped and quantified the areas of aggregation for Iba1+ cells and GFAP+ cells near the injury track. The results indicated a significant increase in Iba1+ cells aggregation near the injury track in the Arid1a cKO group after TBI, reaching peak levels at 7 dpi and persisting until 42 dpi, the longest timepoint we tested (Fig. 2c). Additionally, we found that more astrocytes accumulated around lesion sites in the Arid1a cKO group compared to WT group (Fig. 2d). Meanwhile, we assessed neuronal density at 7 dpi and found that Arid1a cKO mice exhibited more severe neuronal loss compared to WT mice (Fig. S5a-b). This finding suggests that the absence of microglial Arid1a exacerbates brain damage during TBI. Taken together, these data indicate that Arid1a ablation in microglia aggravates glial scar formation and brain damage following TBI, underscoring the essential role of ARID1A in regulating microglial responses to brain injury.
Increased microglial migration in Arid1a cKO mice following TBITo determine whether the increased number of microglia in the cKO-TBI group is due to enhanced migration and/or proliferation of microglia with Aridla-deficiency under TBI conditions, we firstly quantified numbers of microglia at intervals of 100 μm along the 300 μm distance away from the injury track at 7 dpi. The results revealed a reduced amount of microglia from proximal to distal locations and a notable increase in microglia accumulation at each distance away from injury track in the Aridla cKO group compared to the WT group. (Fig. 3a-b).
Fig. 3
Selective deletion of Arid1a in microglia results in increased microglial migration. a Representative image of Iba1 (red) staining around the injury sites at 7 dpi. The injury track is indicated by the yellow dotted lines. The distance between adjacent dotted lines is 100 μm. Scale bar: 50 μm. b Quantification of the density of Iba1+ cells at 7 dpi. n = 4 mice per group. c-d Representative images (c) and quantification (d) of Iba1 (red) and BrdU (green) immunostaining around the injury sites at 7 dpi. Scale bar: 50 μm. n = 4 mice per group. e Schematic diagram of the transwell assay for primary cultured microglia. f-g Transwell migration assay measuring primary microglia migration. cKO microglia exhibited increased migratory capability compared with WT group. n = 4. Scale bar: 100 μm. Data are presented as mean ± SEM; ns, no significance; *P < 0.05 by Student’s t-test
In order to compare the proliferative capacity between WT and Aridla cKO microglia, we administered intraperitoneal injections of BrdU to WT and cKO mice at 30 min before TBI. We counted the BrdU+ Iba1+ cells with a specific focus on regions adjacent to the glial scar at 7 dpi. Surprisingly, the quantification results demonstrated that there was no significant difference in BrdU+Iba1+ cells between the Aridla cKO and WT groups after TBI (Fig. 3c-d), suggesting that the increased number of microglia in the cKO-TBI group is not caused by the enhanced proliferation of Aridla cKO microglia.
Next, we isolated primary microglia from neonatal pups and performed a transwell migration assay in vitro (Fig. 3e). The results showed that there were more Aridla cKO microglia migrated to the lower chamber when compared to WT microglia (Fig. 3f-g), supporting that Arid1a cKO microglia exhibit enhanced migratory capacity compared to WT control.
To rule out the influence of macrophages, we performed immunofluorescence co-staining using Tmem119 alongside Iba1. The results showed that the majority of Tmem119+/Iba1+ cells around the injury site post-TBI were also Iba1+ microglia, with no significant differences observed between the WT and cKO groups (Fig. S6a-b). These findings suggested that macrophage infiltration did not significantly impact microglial accumulation following TBI. Altogether, these data indicate that the loss of Arid1a in microglia enhances their migratory tendency, leading to more severe gliosis after TBI.
Enhanced microglia response to injury in Arid1a cKO miceSince microglia plays a pivotal role in neuroinflammation after TBI [32, 34] and our previous research demonstrates that Arid1a-deficient microglia exhibit an enhanced neuroinflammatory response during the developmental stage [25], we then explored whether Aridla loss-of-function could affect the inflammatory responses in microglia following TBI. qRT-PCR analysis showed that the mRNA expression levels of pro-inflammatory cytokines (Il6, TNFα, Il1β) increased, but the anti-inflammatory cytokine Arg1 decreased in the lesion tissues of Aridla cKO mice at 7 dpi (Fig. 4a-d). Consistently, there was a significant increase in the percentage of Iba1+CD16/32+ microglia and a decrease in the percentage of Iba1+CD206+ microglia among Iba1+ cells around the lesion sites in Aridla cKO mice compared to that in WT mice (Fig. 4e-g). These results support that Aridla cKO microglia surrounding the glial scar exhibit a higher-grade pro-inflammatory state after TBI. To further confirm the results, we performed a flow cytometric analysis with the injured brain tissue at 7 dpi. Again, our results validated those microglia (CD11b+CD45−) in the Aridla cKO group at the injury site exhibited a higher proportion of pro-inflammatory state (CD16/32+) and a lower proportion of anti-inflammatory or tissue-repairing state (CD206+) compared to microglia in the WT group (Fig. 4h-j).
Fig. 4
Increased microglial response to injury in Arid1a cKO mice following TBI. a-d The mRNA expression levels of proinflammatory cytokines, Il1β(a), Il6(b), TNFα(c) and anti-inflammatory cytokine Arg1(d) in brain tissues at 7 dpi. n = 4. e Representative images of Iba1, CD16/32 and CD206 immunostaining of brain sections at 7 dpi. f, g Quantification of microglia at proinflammatory state (f) or anti-inflammatory state (g). n = 3. h-j Representative images (h) and quantifications of flow cytometric analysis of CD16/32+ microglia (i) and CD206+ microglia (j). n = 3 or 4. Data are presented as mean ± SEM; *P < 0.05; **P < 0.01; ***P < 0.001 by Student’s t-test
We next applied ATP and LPS to activate microglia from homeostatic state to activated phenotype in vitro [25], and detected a higher expression of pro-inflammatory cytokines in Aridla cKO microglia with LPS stimulation compared to that with ATP stimulation (Fig. S7a-c). Therefore, we adopted the LPS stimulation protocol for subsequent experiments. Consistent with the in vivo results, the cKO primary microglia subjected to LPS stimulation in vitro did exhibit a higher proportion of pro-inflammatory state (Fig. S7a-f), supporting the enhanced neuroinflammatory response to injury in microglial Arid1a-depleted mice.
ARID1A regulates the injury-induced reactivity of microglia by repressing Ccl5To gain mechanistic insights into how ARID1A regulates microglial migration and reactivity following TBI, we conducted an in-depth transcriptomic comparison between Arid1a cKO and WT microglia. Microglia (CD11b+CD45−) were scrupulously harvested using flow cytometry from the areas adjacent to the TBI lesions in mice from both WT and cKO groups [25]. To assess the similarity between the WT and cKO samples, we conducted Pearson correlation analysis and principal components analysis (PCA) on the transcriptomes. The Pearson correlation coefficients indicated strong positive correlations between the gene expression profiles of the two groups. In PCA, PC1 and PC2 explained 54% and 20% of the variance, respectively. These analyses demonstrated that our samples exhibited significant inter-group differences and intra-group consistency (Fig. S8a-b). The RNA sequencing revealed significant changes in gene expression in Arid1a cKO microglia post-TBI (p-value < 0.01; |log2 fold change| > 1) (Fig. 5a). To understand the roles of these up- and down-regulated differentially expressed genes (DEGs), we performed Gene Ontology (GO) analysis and found that up-regulated DEGs were significantly associated with migration- and inflammatory response-relevant GO terms, such as Ccl5, Il17ra and C5ar1 (Fig. 5b-c).
Fig. 5
ARID1A directly regulates Ccl5. a Heatmap showing DEGs in Arid1a cKO microglia post-TBI. n = 3. b Top significantly enriched GO terms of up-regulated and down-regulated genes post-TBI. Up-regulated DEGs are significantly associated with GO terms related to migration and inflammatory response. c Volcano plot for DEGs with log2 fold change. Key DEGs of interest, including Ccl5, Il17ra and C5ar1, are highlighted within the black boxes. d Genome-browser view of Ccl5 from the RNA-seq, ChIP-seq, and ATAC-seq datasets. The black dotted rectangle highlights the peak gain. e-f Both qPCR (e) and ELISA (f) assays reveal a significant upregulation of Ccl5 expression in cKO microglia compared to WT group in vitro. n = 4. Data are presented as mean ± SEM; *P < 0.05; **P < 0.01 by Student’s t-test
CCL5 is a well-known chemokine that plays a crucial role in promoting migration of various cells. To investigate whether ARID1A directly regulates Ccl5, we re-analyzed publicly available ChIP-seq and ATAC-seq databases related with ARID1A [25, 35]. We found that ARID1A directly binds the coding regions of the Ccl5 gene, the chromatin accessibility is significantly increased at the Ccl5 loci after Arid1a deletion (Fig. 5d), This supports the idea that Ccl5 may be directly regulated by ARID1A, rather than Il17ra and C5ar1(Fig. S9a-b). To experimentally validate Ccl5 as a downstream target of ARID1A, we isolated microglia from WT and cKO newborn pups, and stimulated them with LPS. The RT-PCR results showed that there was a significant up-regulation of Ccl5 mRNA expression in Arid1a cKO microglia after LPS stimulation (Fig. 5e). Our ELISA assay also detected an elevated CCL5 protein level in cell culture supernatant of Arid1a cKO microglia in response to LPS stimulation (Fig. 5f). These findings support that Ccl5 expression was repressed by ARID1A.
To investigate the potential of Ccl5 in regulating the migration of microglia, we administrated anti-CCL5 neutralizing antibody to treat primary microglia and observed a reduction in the migration capability to the lower chamber (Fig. 6a-b) as well as a phonotypic switching from pro-inflammatory state to tissue-repairing state in cKO microglia (Fig. 6c-d). These results suggest that blocking CCL5 could rescue the migration and pro-inflammation defects in Arid1a cKO microglia.
Fig. 6
Arid1a regulates microglial migration and state through Ccl5. a-b Representative images (a) and quantification (b) of microglia migration in transwell assay following treatment with anti-CCL5 antibody. n = 4. Scale bar: 100 μm. c-e Representative images (c) and quantification of flow cytometry in vitro. Quantification of flow cytometric analysis of CD16/32+ microglia (d) and CD206+ microglia (e). n = 4. Data are presented as mean ± SEM; ns, no significance; *P < 0.05; **P < 0.01 by Student’s t-test
Auraptene rescues abnormal microglia migration and reactivity in Arid1a cKO mice after TBIRecent studies have reported that AUR exerts anti-inflammatory effects as evidenced by suppressing inflammatory responses in the ischemic brain and by ameliorating LPS-induced inflammation in the mouse brain [36,37,38]. Since 0.2 μM AUR can significantly decrease the secretion of CCL5 secreted by LPS-stimulated oral epithelial cells [39], we speculated that AUR might be beneficial to Arid1a cKO mice after TBI. To test this hypothesis, we subcutaneously administered AUR at a dosage of 25 mg/kg/day for 3 consecutive days before TBI and for 7 consecutive days after TBI. Such a dosage of AUR can efficiently act as an anti-inflammatory agent in the mouse brain with ischemic surgery [28]. We then performed IF staining and observed that AUR treatment significantly reduced the accumulation of microglia at lesion sites in the cKO group at 7 dpi (Fig. 7a-b). Similarly, the aggregation of astrocytes in the cKO group was also significantly reduced following AUR treatment (Fig. 7a and c). Subsequent analyses revealed a notable decrease in the density of microglia near the injury site in the cKO group following AUR treatment (Fig. 7d-e). Consistently, transwell migration assay proved that treatment of AUR did reduce the migration of Arid1a cKO microglia (Fig. S10a-b). Additionally, we observed that AUR treatment effectively attenuated the percentage of pro-inflammatory state (CD16/32+ microglia) from 28% down to 19% in Arid1a cKO primary microglia (Fig. 7f-h).
Fig. 7
Auraptene treatment alleviates the formation of glial scar in Arid1a cKO mice following TBI. a Representative image of IF staining for Iba1(red), GFAP (green) and nuclei (DAPI, blue) in the auraptene-treated brains at 7 dpi. Scale bars: left panel, 1,000 μm; right panel, 100 μm. b, c Quantification of Iba1+ (b) and GFAP+ area (c) at 7 dpi. n = 3. d Representative images of Iba1 (red) immunostaining around the injury sites at 7 dpi. The injury tracks are indicated by the yellow lines. n = 3. Scale bar: 50 μm. e Quantification of the density of Iba1+ cells around the lesion sites at 7 dpi. f-h AUR represses inflammatory response of Arid1a cKO microglia upon LPS-stimulation. Representative images of flow cytometric analysis in vitro (f). Quantification of CD16/32+ microglia (g) and CD206+ microglia (h) in the flow cytometric assay. n = 3 or 4. Data are presented as mean ± SEM; ns, no significance; *P < 0.05; **P < 0.01; ***P < 0.001
Altogether, this study provides evidence that Arid1a plays a crucial role in regulating microglial migration and reactivity in response to TBI. Microglial ARID1A deficiency leads to exacerbated glial scar formation and elevated inflammation through the upregulation of CCL5. Importantly, treatment with AUR ameliorates the deficits of Arid1a cKO mice after TBI.
留言 (0)