Cell culture, treatments, and transfection
A431 (human epidermoid cell line; ATCC# CCL-1555) and A549 (human lung carcinoma cell line; ATCC# CCL-185) were kindly provided by Dr. Ada Sacchi (Istituto Nazionale Tumori Regina Elena, Rome, Italy), H1299 (human lung carcinoma cell line; ATCC# CRL-5803) was kindly provided by Dr. Giovanni Blandino (Istituto Nazionale Tumori Regina Elena, Rome, Italy), U-2 OS (human osteosarcoma cell line; ATCC# HTB-96) was kindly provided by Prof. Francesco Blasi (Università degli Studi di Milano, Milan, Italy), and HaCaT (human keratinocyte cell line) was kindly provided by Prof. Antonio Costanzo (Humanitas Research Hospital, Milan, Italy). All cells were maintained in DMEM enriched with 10% fetal bovine serum (Euroclone #ECS0165L), 1 mM l-glutamine (Euroclone #ECB3000D), 100 units/mL penicillin, and 100 μg/mL streptomycin (Euroclone #ECB3001D) at 37 °C in a humidified atmosphere of 5% (v/v) CO2. Cells were maintained in culture for no more than ten passages and underwent routine testing to ensure that they are mycoplasma-free. For treatments with thalidomide (Tocris #50–35-1) and lenalidomide (LGC products #191,732–72-6), 5 × 104 cells were seeded onto 24-well multi-plates and 20 h later incubated with different drug concentrations and times indicated in the figure legends. For transient transfection, 5 × 104 A431 cells were plated in 24-well multi-plates and on the next day transfected with Lipofectamine 2000 (Invitrogen #11,668,019) with increasing amount of CEREBLON (CRBN) encoding plasmid or with four different p63-small hairpin RNA (sh-p63) vectors with sequence homology to four different regions of p63 mRNA (OriGene Technologies # TF308688); shRNA-SCRAMBLED (sh-SCRB) was used as a control. For stable transfection of A431 cells, shp63#3/shp63#4 and shSCRB plasmids were transfected with Lipofectamine 2000 (Invitrogen #11668019), and after 2 days, 1 μg/mL puromycin was added to select stable transfectants. The cells were maintained as polyclonal populations under puromycin at 0.25 μg/mL. For U-2 OS, 5 × 104 cells were plated in 24-well multi-plates and the next day transfected with Lipofectamine 2000 with increasing amount of ΔNp63α encoding plasmid.
Western blot (WB) analysis
At the indicated times, cells were lysed in 100 μL of Loading Buffer 2X (2% sodium dodecyl sulfate, 30% glycerol, 144 mM β-mercaptoethanol, 100 mM Tris–HCl pH 6.8, and 0.1% bromophenol blue). Samples were incubated at 98 °C for 10 min and resolved by SDS-PAGE. Proteins were transferred to a nitrocellulose membrane (Amersham #GEH10600001). The blots were incubated with the following antibodies (Abs): anti-p63 4A4 (Santa Cruz Biotechnology sc-8431), anti-ACE2 (Abcam ab15348), anti-CRBN (Cell Signaling Technology D8H3S), and anti-actin (Santa Cruz Biotechnology sc-8432). The following secondary Abs were used: goat anti-mouse IgG-HRP (Santa Cruz Biotechnology sc-2005) and goat anti-rabbit IgG-HRP (Santa Cruz Biotechnology sc-2030). Proteins were visualized by an enhanced chemiluminescence method (GeneSpin #STSE500) according to the manufacturer’s instructions using a ChemiDoc Touch (BioRad).
Plasmids
The plasmids carrying ΔNp63α, CRBN, or shCRBN were previously described [17].
RNA purification, reverse transcriptase RT-PCR, and quantitative real-time qPCR analyses
Total mRNA from cells was isolated using the RNeasy miniKit (Qiagen #74,104). cDNA was synthesized by M-MLV RTase and amplified with GoTaq DNA polymerase (Promega #M3001). For quantitative PCR analysis, mRNA expression level was evaluated using the Power SYBR Green PCR Master Mix with ABI Prism 7500HT Fast Real-Time PCR System Detector (Applied Biosystems). Relative mRNA expression levels were determined by using the 2-∆∆CT method, employing GAPDH gene expression for data normalization. All reactions were performed in triplicate. Primer sequences are as follows:
ACE2 forward 5′-CAT TGG AGC AAG TGT TGG ATC TT-3′;
ACE2 reverse 5′-GAG CTA ATG CAT GCC ATT CTC A-3′;
GAPDH forward 5′-TCC CTG AGC TGA ACG GGA AG-3′;
GAPDH reverse 5′-GGA GGA GTG GGT GTC GCT GT-3′.
ELISA assay
HaCaT cells, 1.8 × 105, were plated in 35-mm culture dishes and treated with recombinant human TNF-α (rhTNF-α; BioLegend # BMS301) 5 ng/mL for 7 or 17 h or with 0.5 µg/mL LPS from E. coli (SIGMA #O111:B4) for 4, 6, or 17 h. Quantification of IL-8 in the supernatants of treated HaCaT cells was performed by ELISA (hIL-8; ImmunoTools #31,670,089) according to the manufacturer’s instructions. The ELISA plates were read by a microplate reader (SAFAS MP96).
Chromatin immunoprecipitation (ChIP)
A431 cells, 4 × 106, were plated in 15-cm culture dishes and treated with LPS 0.5 µg/mL for 2 h. Then, proteins were cross-linked to DNA in living nuclei and the ChIP assay was performed using the MAGnify™ Chromatin Immunoprecipitation System (Thermo Fisher #492,024), as described by the manufacturer. The following quantities of Abs were used to immunoprecipitate the DNA–protein complexes: 2 μg of anti-p63α (D2K8X) XP® Rabbit mAb (Cell Signaling Technology #131,095), 2 μg of anti-acetylated H4-histone Ab (BioRad #AHP148), and 1 μg of an unrelated, negative control Ab. DNA fragments obtained by the ChIP assay were analyzed by qPCR using specific primers spanning the responsive elements (RE) found in the first intron of the ACE2 gene. Primers specific for exon 8 of the ACE2 gene were used as the negative control. As a positive control for PCR and as normalizer, DNA prepared from samples prior to immunoprecipitation (whole cell lysates) was used as total or input DNA. Primer sequences are as follows:
First intron of the ACE2 gene (+ 1067; + 1087):
Forward: 5′-ACGACGCGTGTGGAGAAGTCCATCAGA-3′;
Reverse: 5′-ACGAGATCTGGTCAACCACACATACCA-3′;
Exon 8 ACE2 gene (+ 19,634; + 19,730):
Forward: 5′-GGGATGCACAGAGAATATTCAAGG-3′;
Reverse: 5′-AGACTGCTTTCTGAACATTTCCTG-3′.
In vitro SARS-CoV-2 spike protein pseudovirus infection
A baculovirus expressing the green fluorescent protein (GFP) and pseudotyped with SARS-CoV-2 spike protein (Montana Molecular #C1110G) was employed to evaluate its entry into cells through ACE2 receptor. To assess Thal-induced effect, 5 × 103 A431 cells were plated in triplicates in 96-well plates. Thal was added 17 h post cell seeding; after 24 h of treatments, 50 μL of pseudo-SARS-CoV-2 suspension (viral titer: 2 × 1010 viral genes (VG) per mL) was added to each well following the manufacturer’s instructions. At 30 h post-infection, cells were washed with PBS, fixed with 4% formaldehyde in PBS for 10 min at room temperature, incubated for 10 min with the reagent containing 4,6-diamidino-2-phenylindole (DAPI), and washed again with PBS. Images were acquired using a Nikon CSU-W1 microscope in widefield mode, using a × 20 objective and analyzed with FIJI [25].
FACS analysis
For FACS analysis, shp63 and shSCRB stable transfectants were plated and infected with the SARS-CoV-2 spike protein pseudovirus as above, and after 24 h, the FACS analysis was performed by a BD (Franklin Lakes, NJ, USA) FACSCanto II flow cytometer. Data were analyzed with FlowJo software (version 10.4.2; BD, Franklin Lakes, NJ, USA).


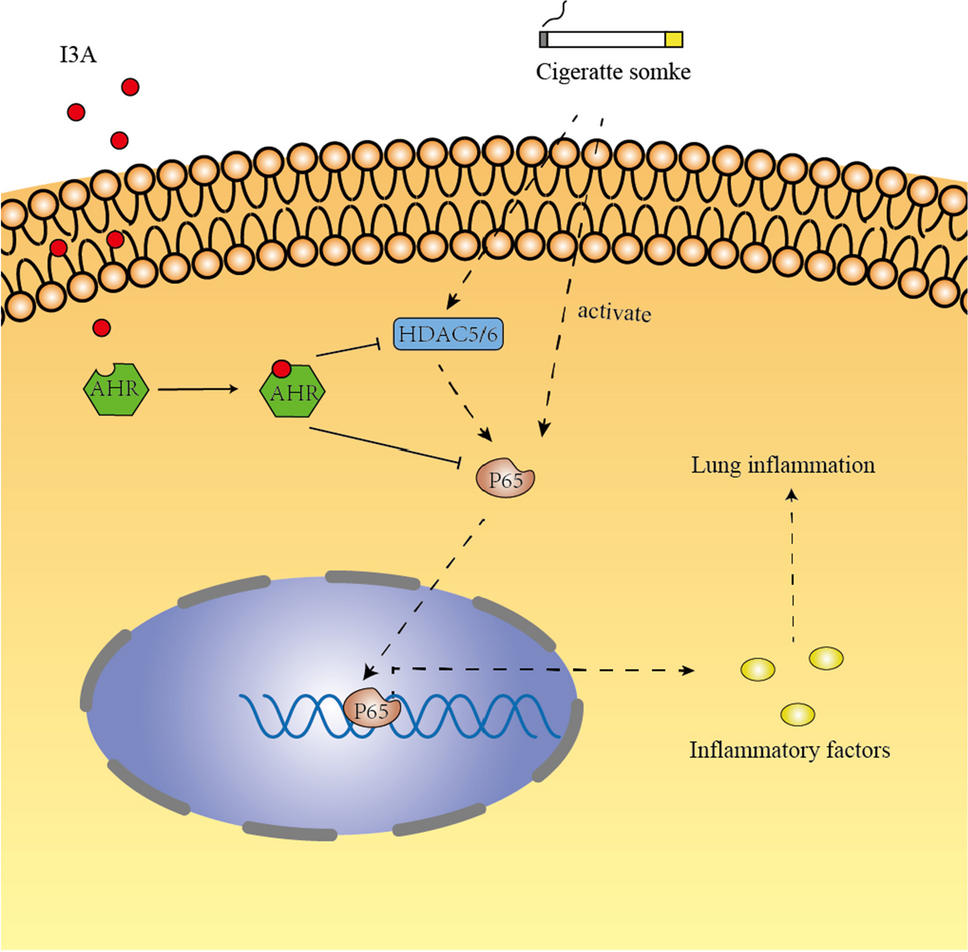
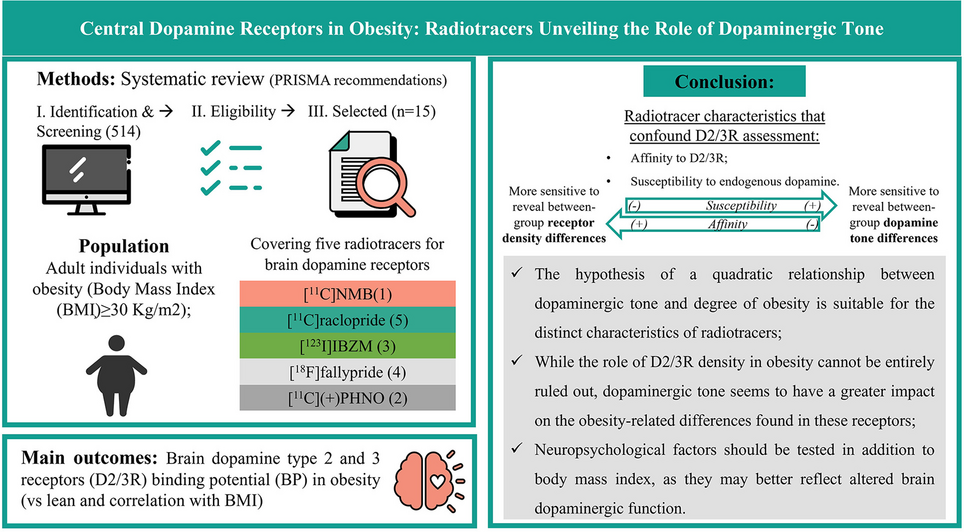
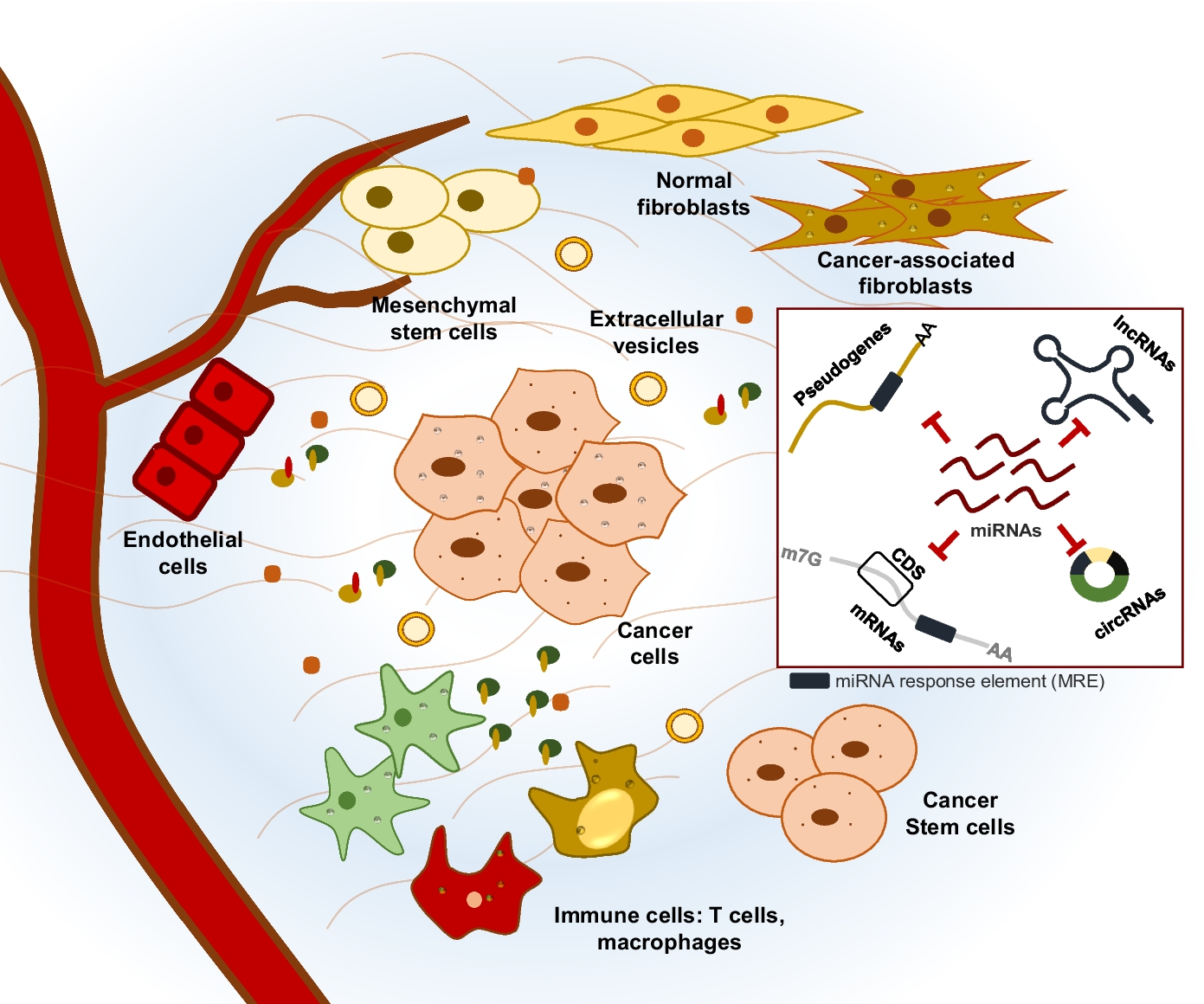
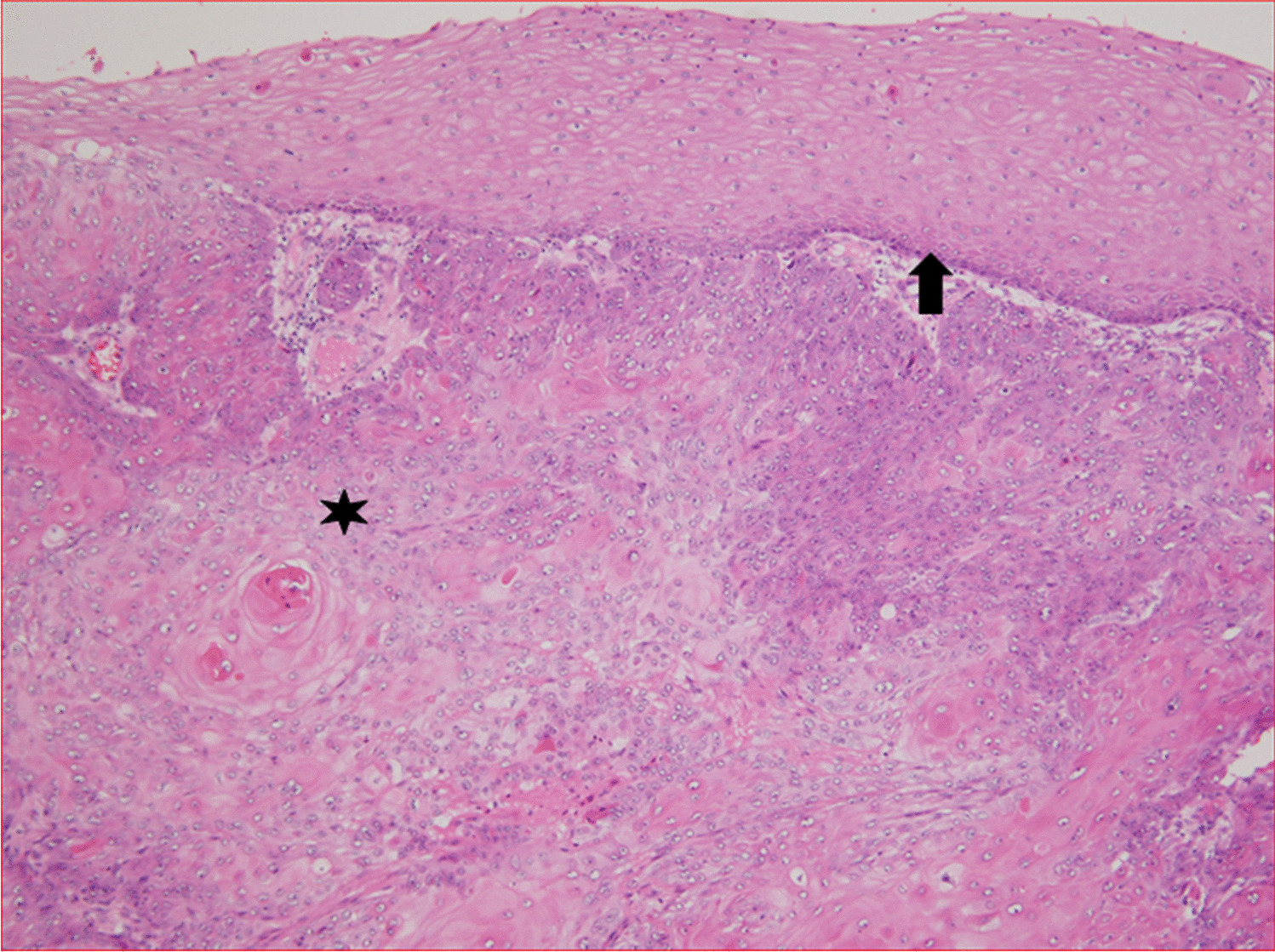



留言 (0)