
記住我




Patient 1 is a female born from the first pregnancy to healthy parents of Romani origin at 34 weeks of gestation, with birth weight and length in the 50th percentile. In her first week of life, she presented with frequent watery stools leading to severe dehydration accompanied by mild metabolic acidosis and hypoglycemia requiring parenteral nutrition. Apart from electrolyte disturbances caused by dehydration, her laboratory findings showed progressive conjugated hyperbilirubinemia, which later resolved spontaneously. Liver enzymes were only transiently elevated. Despite all the therapeutic efforts, her clinical status progressively deteriorated, and she died of septic shock at 18 weeks of age (Table 1).
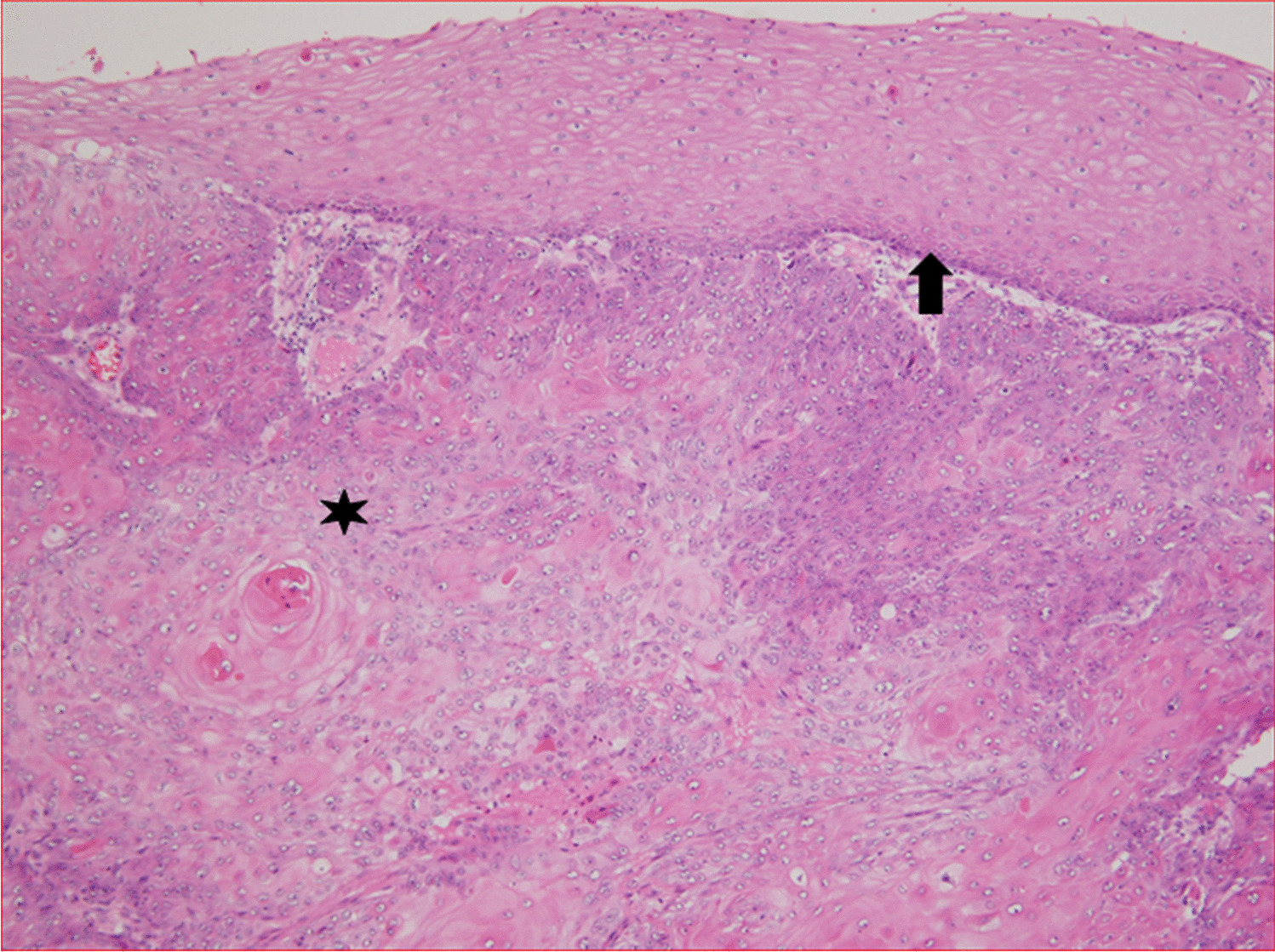
Patient 2 is a male born from the 12th pregnancy and 7th parity to another couple of healthy parents of Romani origin at 37 weeks of gestation, with birth weight and length in the 22nd percentile. Four days after birth, he presented with frequent watery acholic stools and malabsorption, leading to severe dehydration requiring parenteral nutrition. Histopathological findings in the small bowel and colon were similar, revealing mild to moderate chronic focally active enteritis and colitis with the presence of numerous apoptotic bodies in the crypt epithelium and disrupted mucosal architecture, including distorted crypts and shortened villi. Unlike patient 1, the symptoms of his enteropathy gradually subsided over time, and by the end of the second year of life, he was switched to full enteral nutrition. This patient also showed signs of renal disease characterized by progressive proteinuria with dominating tubular component (up to 205 mg/mmol creatinine), and mild hepatopathy (intermittent elevation of liver enzymes with preserved function of protein synthesis and elimination) (Table 1). He died at the age of 5.5 years due to aspiration in home care.
Table 1 Clinical characteristics of patients with the AP1S1 c.269 T > C variantBoth patients had ichthyosis and keratoderma notable already in the neonatal period. Otoacoustic emissions repeatedly failed to be detected in both patients and patient 2 later showed clinical signs of deafness. Neurological symptoms, characteristic of MEDNIK syndrome, were not initially observed. However, patient 2 subsequently exhibited global developmental delay, intellectual disability, and peripheral neuropathy. Characteristic disturbances in copper metabolism were also present in the patients (Table 1). Both patients experienced several episodes of sepsis; the majority of them were linked to re-initiation of enteral nutrition. The spectrum of pathogens detected in blood cultures suggested either catheter-related infections or translocation of bacteria from the intestine. Furthermore, both patients suffered from recurrent venous thrombosis. The second patient also suffered from hypogammaglobulinemia requiring repeated immunoglobulin substitution.
Genetic analysisWhole exome sequencing (WES) was performed in both patients independently, and the results did not reveal any known monogenic enteropathy, inborn error of immunity, or metabolic disorder. Analysis of homozygous variants revealed a missense c.269 T > C (p.L90P) variant in the AP1S1 gene (Fig. 1b) predicted to be highly deleterious (CADD: 28.5) and leading to a change of a highly conserved leucine residue (Supplementary Fig. 1a). This variant had not been reported at the time of our initial analysis but was later shown to be pathogenic by Klee et al. [13]. Parents and two siblings of patient 1 and the mother of patient 2 were found to be heterozygous carriers of the c.269 T > C variant by Sanger sequencing (Fig. 1b, c); the father of patient 2 did not give consent to genetic examination.
Of note, most of the previously described cases with AP1S1 homozygous variants were from consanguineous couples. Although both families of our patients denied consanguinity, homozygosity mapping analysis of the WES data showed large runs of homozygosity in both patients 1 (140.13 Mb) and 2 (362.13 Mb), which is highly suggestive of parental blood relationships (Supplementary Fig. 1b). Furthermore, examination of the coding SNVs in the region surrounding the variant in both patients revealed an identical haplotype over a 300 kb-long region flanking the variant. The genotype of the patients in this region consists mostly of minor alleles with a population frequency of 0.08–4.8%, which suggests that the c.269 T > C variant is linked with this specific haplotype and has a founder nature.
Molecular and functional characterization of AP-1 σ1AL90PTo assess the impact of the L90P variant on σ1A and the AP-1 complex we used a heterologous expression system involving transient expression of WT and variant σ1A cDNA constructs in human HeLa, HEK293T and HAP1 cells.
Decreased levels and assembly of σ1AL90PFirst, we examined the levels of the σ1AWT and σ1AL90P proteins appended with a C-terminal triple-myc tag in transfected HeLa cells [20]. We observed that σ1AL90P was expressed at ~ 56% the levels of σ1AWT (Fig. 2a), possibly due to destabilization and partial degradation of the protein. This effect is consistent with the expected disruption of the α-helix harboring leucine-90 by the conformationally restricted proline residue (Supplementary Fig. 1a). Next, we analyzed the impact of the L90P variant on the assembly of σ1A into the AP-1 complex. Since the σ1A subunit interacts most extensively with the γ1 subunit [21], we examined the effect of the L90P variant on the assembly of σ1A with γ1. To this end, we transfected HEK293T cells with plasmids encoding myc-tagged σ1AWT or σ1AL90P, and examined the assembly of these constructs with endogenous γ1. Reciprocal co-immunoprecipitation experiments showed that assembly of γ1 with σ1AL90P was reduced to 0–13% relative to σ1AWT (Fig. 2b, first and fourth blots from top). The fact that the decrease in assembly was greater than the decrease in protein levels (Fig. 2a top blot and Fig. 2b bottom blot) indicated that the L90P substitution also impairs the interaction of σ1A with γ1, thus preventing proper assembly of the AP-1 complex.
Fig.2
The AP-1 σ1A L90P substitution impairs assembly of the AP-1 complex and recognition of dileucine signals. a Decreased expression of myc-tagged σ1A L90P relative to WT myc-tagged σ1A expressed by transient transfection in HeLa cells and analyzed by SDS-PAGE and immunoblotting (IB). Blots of endogenous γ1, μ1 and β-tubulin are included as loading controls. b Impaired assembly of myc-tagged σ1A L90P into the AP-1 complex. HEK293T cells were transiently transfected with plasmids encoding either WT or L90P myc-tagged σ1A, and cell extracts were subjected to immunoprecipitation (IP) with anti-myc followed by SDS-PAGE and IB with anti-γ1 or anti-myc, or IP with anti-γ1 followed by SDS-PAGE and IB with anti-γ1 or anti-myc. Untransfected cells (-) were used as control. Notice that both permutations of IP and IB showed decreased co-immunoprecipitation of endogenous γ1 with myc-tagged σ1A L90P relative to myc-tagged WT σ1A (ranging from 0 to 13%, depending on the antibody combination; first and fourth blots from top). c Y3H assays showing lack of interaction of σ1A L90P with dileucine-based sorting signals. The AP-1 γ1, AP-2 αC and AP-3 δ subunits were subcloned in the Gal4 transcriptional activation domain (AD) vector pGADT7. The cytosolic tails of LIMP-II or tyrosinase and the indicated σ subunits were subcloned in the MCS1 and MCS2 of the Gal4 DNA binding domain (BD) vector pBridge, respectively. Transformants were plated on medium lacking leucine, tryptophan and methionine but containing histidine (+ His, bottom panel) to control for viability and loading, and on the same medium lacking histidine (-His, top panels) to detect protein interactions. The top panel shows the interaction of the γ1-σ1A hemicomplex with the dileucine motifs in the cytosolic tails of LIMP-II and tyrosinase (ERAPLI and ERQPLL, respectively [6]; and that the σ1A L90P substitution abrogates this interaction. Note the selective interaction of the LIMP-II and tyrosinase tails with the AP-1 γ1-σ1A hemicomplex but not with mismatched combinations of AP subunits. Additional controls in the assay include the interaction of the LIMP-II tail with AP-2 αC-σ2 and of the tyrosinase tail with AP-3 δ-σ3A (but not with mismatched combinations of AP subunits). Yeast co-transformation of pBridge-based constructs with a Gal4 AD-SV40 T-Ag fusion construct and of AD-AP subunit fusions with a Gal4 BD-p53 construct were used as negative controls. Co-transformants co-expressing AD-SV40 T-Ag and BD-p53 fusions provided a positive control for interactions
Abrogation of [DE]XXXL[LI] motif recognition by σ1AL90PNext, we used a yeast three-hybrid assay (Y3H) to examine the impact of the σ1A L90P variant on the recognition of dileucine-based, [DE]XXXL[LI] sorting motifs, which bind to a site including residues from both σ1A and γ1 [4, 6, 21]. The assays were performed by co-expression of different combinations of large and small subunits of AP-1, AP-2 and AP-3 with the [DE]XXXL[LI]-containing cytosolic tails of the lysosomal membrane protein LIMP-II and the melanosomal membrane protein tyrosinase [4, 6] (Fig. 2c). These experiments showed that σ1AWT, in combination with γ1 but not with the homologous AP-2 αC and AP-3 δ subunits, interacted with the cytosolic tail of LIMP-II and tyrosinase (Fig. 2c). Importantly, we observed that the L90P substitution completely abolished the interaction of the γ1-σ1A hemicomplex with the LIMP-II and tyrosinase tails in the Y3H assay (Fig. 2c). This result was consistent with the virtual inability of σ1A to assemble with γ1 (Fig. 2b). Because of the proximity of the L90P substitution to the [DE]XXXL[LI]-binding site (Supplementary Fig. 1a), this variant may also impair the ability of σ1A to participate in signal recognition.
Localization of the AP-1 complex in σ1AL90P-expressing cellsNext, we investigated the effect of the σ1A L90P substitution on the intracellular localization of AP-1 by rescue of HAP1 cells having knock-out (KO) of all three σ1 isoforms (σ1A, σ1B and σ1C). We observed that KO of all three σ1 isoforms markedly reduced the levels of the γ1 and μ1A proteins, as analyzed by immunoblotting (Supplementary Fig. 2a, b). Expression of myc-tagged σ1AWT in these cells resulted in partial restoration of endogenous γ1 levels, whereas expression of myc-tagged σ1AL90P led to minimal or no recovery relative to untransfected cells (Fig. 3a). We also examined the effect of σ1AL90P expression on the rescue of AP-1 by immunofluorescence microscopy. Triple KO of σ1 isoforms dramatically reduced TGN/endosomal staining for γ1 (Fig. 3b). Expression of myc-tagged σ1AWT rescued TGN/endosomal γ1 staining, whereas expression of myc-tagged σ1AL90P did not (Fig. 3c). These data indicated that the σ1A L90P substitution disrupts the association of γ1 with TGN/endosomes, likely due to impaired assembly of the whole AP-1 complex.
Fig.3
The σ1A L90P substitution prevents rescue of the association of AP-1 with TGN/endosomes in triple σ1-KO cells. a IB analysis of cell lysates from WT HAP1 cells and triple σ1-KO HAP1 cells untransfected (-) or transfected with plasmids encoding myc-tagged WT or L90P σ1A. Notice the partial rescue of γ1 levels in triple σ1-KO cells expressing σ1WT but not L90P σ1A. IB with anti-β-tubulin is shown as loading control. b Confocal immunofluorescence microscopy of WT and triple σ1-KO HAP1 cells stained for endogenous γ1 (red channel) and the TGN marker TGN46 (green channel). Notice the co-localization of γ1 and TGN46 in the perinuclear region of WT HAP1 cells, and the marked reduction in γ1 signal in the triple σ1-KO cells. c Confocal immunofluorescence microscopy of triple σ1-KO HAP1 cells transfected with plasmids encoding myc-tagged WT or L90P σ1A, and stained for endogenous γ1 (red channel) and the myc epitope (green channel). Notice the rescue of perinuclear γ1 immunostaining by expression of WT but not L90P σ1A. In b and c, cell edges are indicated with dashed lines; scale bars: 10 μm
Impact of σ1AL90P on sorting of tight-junction proteinsKlee et al. showed that σ1AL90P-mediated disruption of AP-1 complex function leads to mislocalization of two tight junction proteins, ZO-1 and claudin 3, in a Caco-2-cellular model of the intestinal barrier [13]. However, due to the lack of material, they could not confirm the results on primary patient samples. We evaluated the localization of these two tight-junction proteins in intestinal biopsies of patient 2. Surprisingly, immunohistochemical staining of both ZO-1 and claudin 3 with the same antibodies used by Klee et al. showed correct localization of these proteins to the apical and basolateral plasma membrane, respectively, with no significant difference compared to the staining pattern of healthy controls (Fig. 4a, b).
Fig.4
Immunohistochemical analysis of patient samples. a Immunohistochemical staining of ZO-1 in intestinal biopsy of healthy control and patient 2. Arrows show apical localization of the protein. b Immunohistochemical staining of claudin-3 in intestinal biopsy of healthy control and patient 2. Arrows show basolateral localization of the protein
Since it has been speculated that other σ1 isoforms could partially compensate for the dysfunctional σ1A isoform in vivo, we evaluated mRNA expression of AP1S1, AP1S2 and AP1S3 genes in FFPE samples of patient 2 taken from the duodenum and rectum at three different time points during the course of the disease. We detected expression of all three σ1 isoforms but no significant changes in the expression of the AP1S2 and AP1S3 isoforms in the patient samples compared to the healthy controls (Supplementary Fig. 3).
Taken together, the functional experiments demonstrated that the σ1AL90P variant is virtually unable to assemble into the AP-1 complex, resulting in abrogation of DE]XXXL[LI]-signal recognition and drastic reduction in association of γ1 with the TGN/endosomes.
留言 (0)