Materials
The following reagents and materials were used in this study: Dulbecco’s modified Eagle medium (DMEM), fetal bovine serum (FBS), and trypsin from Gibco-BRL (New York, USA); penicillin and streptomycin from Hyclone (Logan, USA); CUR sourced from the Chinese National Institute for the Control of Pharmaceutical and Biological Products (Beijing, China); MTT assay kit purchased from Abcam (Cambridge, UK); apoptosis, Western blotting, and real-time quantitative polymerase chain reaction (RT-qPCR) assay kits, as well as H2O2 and Brusatol, obtained from Sigma-Aldrich (Saint Louis, USA); Malondialdehyde (MDA), superoxide dismutase (SOD), ROS, and glutathione (GSH) assay kits acquired from Nanjing Jiancheng Bioengineering Institute (Nanjing, China); and Bcl-2, Caspase-3, Bax, Nrf-2, and NQO-1 antibodies sourced from Affinity Biosciences CO (Beijing, China).
Cell culture
PC12 cells were cultured following a previously published protocol [16]. The cells were maintained in DMEM medium from Gibco-BRL (New York, USA), supplemented with 10% FBS and 1% penicillin/streptomycin from Hyclone (Logan, USA). The cell culture was incubated at 37 °C with 5% CO2. Media were refreshed every 48 h, and subculturing was performed when the cell density reached 90%.
Cell treatment
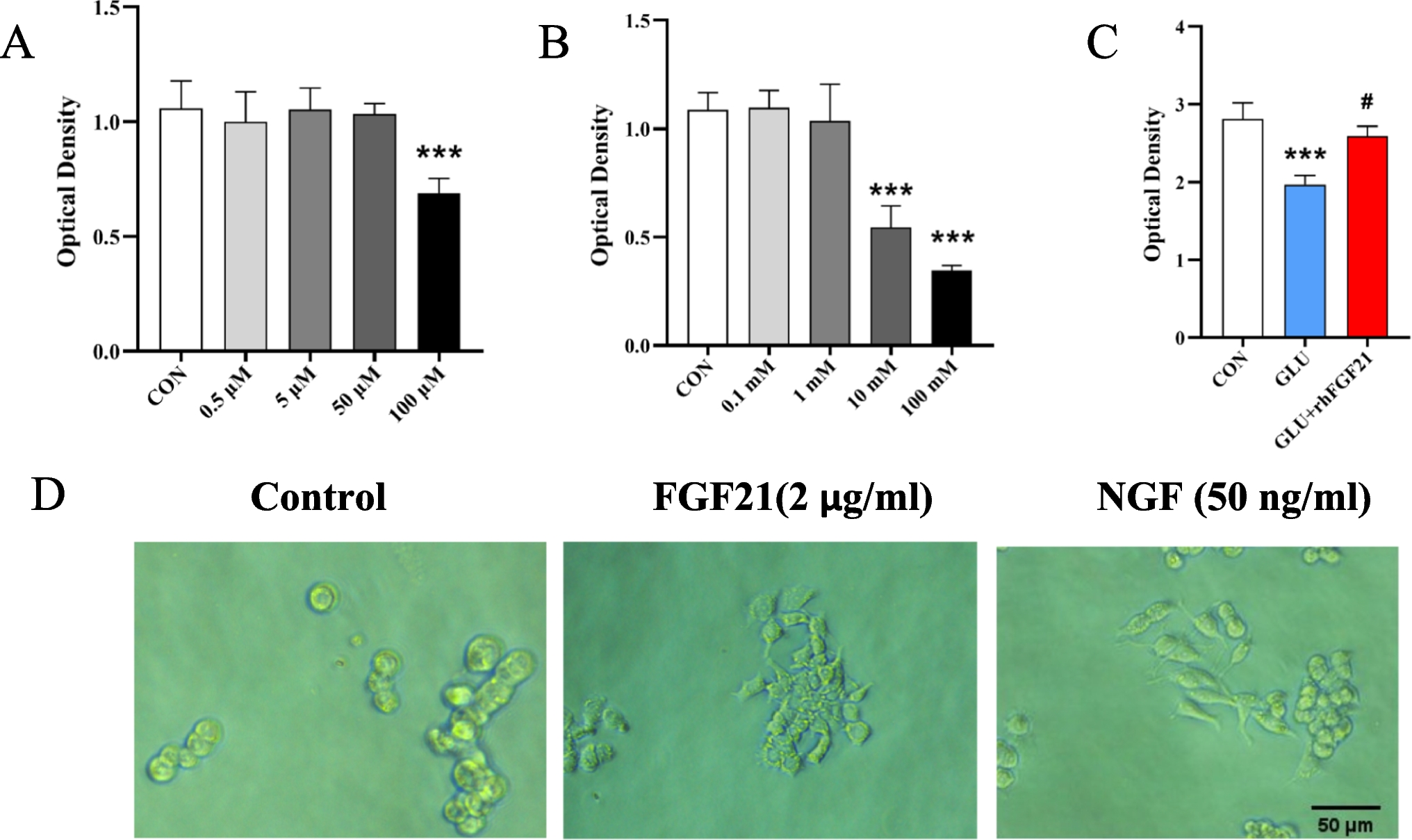
The PC12 cells were divided into five groups: control group, CUR group, H2O2 group, H2O2 + CUR group, and H2O2 + CUR + Brusatol group. CUR was used at a concentration of 3 μM, H2O2 at a concentration of 300 μM, and Brusatol at a concentration of 100 nM for the treatments.
Cell Viability Assay
According to the previously described method, cell viability was assessed using the MTT assay [17]. The cells were divided into 5 groups and seeded at an initial density of 1 × 104 cells per well for 24 h. Two groups were exposed to varying concentrations of H2O2 (100, 200, 300, 400, 500 μM) and CUR (1, 3, 10 μM). One group was pre-treated with different concentrations of CUR for 2 h, followed by 24 h of co-incubation with 300 μM H2O2. The last group was treated with 300 μM H2O2 and incubated for varying durations. After incubation, 10 μL of MTT solution (5 mg/mL) was added to each well and incubated for 3 h at 37 °C. The formazan crystals formed were dissolved in 100 μL of Dimethylsulfoxide (DMSO) per well [18]. The mixture was shaken at room temperature for 15 min, and absorbance was measured at 570 nm using a microplate reader (Bio-Rad, USA) to determine cell viability.
Apoptosis Assay
The apoptosis rate of cells was determined using the Annexin V/propidium iodide (PI) double-staining method and Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay, following established protocols [19, 20]. For Annexin V/PI staining, treated cells were collected, washed, and resuspended in binding buffer containing Annexin V-FITC and PI. After incubating the cells for 15 min, flow cytometry analysis was performed.
The TUNEL assay was performed utilizing the TUNEL apoptosis detection kit, where 4’,6-diamidino-2-phenylindole (DAPI) was used to stain the nuclei. The resulting observations were recorded using fluorescence microscopy (Olympus, Japan).
RT-qPCR Analysis
Total RNA extraction was conducted using the TRIzol reagent following treatment. RT-PCR analysis was carried out using the First-Strand cDNA Synthesis Kit in combination with the PrimeScript RT reagent kit on a SYBR green system (Toyobo, Japan Ltd., Japan). The primer sequences used are detailed in Table 1.
Table 1 Primers used for target amplification in this studyWestern Blotting Analysis
Protein concentrations were determined using the BCA protein assay kit. The proteins were separated on a 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) gel and subsequently transferred to a polyvinylidene fluoride (PVDF) membrane (Millipore, USA). The membrane was blocked overnight at 4 °C with the respective primary antibodies (Bcl-2, Caspase-3, Bax, Nrf-2, NQO-1). Following this, the membrane was washed with tris buffered saline + 0.5% [v/v] Tween-20 (TBST) and incubated for 2 h at room temperature with HRP-conjugated secondary antibodies (1:1000 dilution). Glyceraldehyde phosphate dehydrogenase (GAPDH) at a 1:1000 dilution served as the internal standard. Quantitative densitometric analysis was conducted using the ImageQuant LAS 4000 mini detection system (GE Healthcare, Buckinghamshire, UK), and the results were analyzed using ImageJ software (National Institutes of Health, Bethesda, MD).
Oxidative Stress Assay
For the in vitro test, intracellular ROS levels were assessed using DCFH-DA reagent following the manufacturer’s instructions. Fluorescence images were captured using a fluorescence microscope and analyzed with ImageJ software. Additionally, GSH concentration was determined using a GSH assay reagent.
In the in vivo test, oxidative stress markers including MDA, SOD, and GSH were measured using specific assay kits. Spinal tissue samples were processed according to the manufacturer’s instructions to extract the liquid supernatant for analysis. MDA levels were measured at 532 nm, he activity of SOD was assessed at 550 nm, and GSH levels were quantified at 420 nm using a microplate reader.
Animals
All animal experiments were conducted following approval from the Ethics Committee of Experimental Animal Management at Guangzhou University of Chinese Medicine (approval number: 20220803007). Sprague–Dawley (SD) male rats weighing 200-250 g were obtained from the Experimental Animal Center of Guangzhou University of Chinese Medicine and housed in the animal laboratory. The rats underwent a one-week adaptation period under the following conditions: five rats per cage, ambient temperature maintained at 22–26 °C, a 12-h light–dark cycle, and humidity ranging from 55 to 68%.
Establishment of the Rat Model of SCI
The SCI animal model was induced using Allen’s method [21]. Rats were anesthetized, secured for shaving, and a median incision was made on the back to expose the T9-11 spinous processes and lamina, using T10 as a reference point. The spinous process and lamina of T10 were removed to expose the intact spinal cord underneath. A PinPoint™ precision SCI impactor was used to strike the T10 spinal cord with a velocity of 1.2 m/s, a depth of 1.0 mm, and a contact time of 85 ms. This caused rapid congestion and tissue redness in the T10 region. Transient spasms and convulsions in the tails and hind limbs confirmed successful establishment of the SCI model. Subsequently, hemostasis was achieved and the wound was sutured, followed by iodine application to the sutured skin. In sham-operated rats, the spinal cords were exposed similarly but not subjected to the impact procedure..
Grouping and Treatment of SCI Model
Fifty mature male Sprague–Dawley (SD) rats weighing 200-250 g were randomly divided into four groups, each comprising 30 rats: (1) sham group, (2) SCI model group, (3) SCI + NS group, and (4) SCI + CUR group. One day after successful SCI modeling, the rats received oral treatments once daily for 14 days at a dose of 50 mg/kg. The SCI + NS group was administered 10 mL/kg of normal saline, while the SCI + CUR group received 50 mg/kg of CUR. On day 14 post-treatment, the rats were euthanized for analysis.
Locomotion Recovery Assay
To evaluate the mobility of rats following SCI, inked footprint analysis and the Basso-Beattie-Bresnahan (BBB) locomotion rating scale were used [22, 23]. Rats were allowed 15 min of free activity in an open field on days 3, 7, and 14 post-surgery. The BBB scale, ranging from 0 to 21 points, was used to assess hindlimb locomotion ability (with 0 indicating paralysis and 21 indicating normal mobility). Inked footprint analysis was conducted on day 14 after injury: the rats’ hind paws were coated with dye and they were guided in a straight line across white paper to capture representative images of their gait and coordination.
Tissue Preparation And Preservation
After euthanizing the rats, tissue samples were collected from the lesion site in the spinal cord. One portion of the tissue was promptly frozen in liquid nitrogen for subsequent oxidative stress assays and western blotting analysis. The remaining tissue was decalcified in 10% ethylenediamine tetraacetic acid (EDTA), dried, embedded in paraffin wax, and then sectioned at a thickness of 5 μm. These sections were used for histological staining and immunohistochemical analysis.
Histopathological Staining
The tissue sections were stained with hematoxylin and eosin (HE) for general histology and with cresyl violet for Nissl staining to assess neuronal morphology and density. Histopathological changes and lesions were observed using an inverted microscope (Olympus, Japan).
Immunofluorescence Staining and Analysis
Before antigen retrieval with 0.01 M citric acid (pH 6.0) at 95 °C for 10 min, tissue sections underwent deparaffinization with xylene and hydration through varying alcohol concentrations. Subsequently, the sections were rinsed with 0.01 mol/L phosphate-buffered saline (PBS) and incubated in a 10% goat serum antibody blocking solution for 2 h at room temperature. Following blocking, the tissues were incubated overnight at 4 °C with primary antibodies against MAP2 (1:200, Boster Biological Engineering Co.), GFAP (1:200, Boster Biological Engineering Co.), and NeuN (1:200, Boster Biological Engineering Co.). The next day, the tissue sections were incubated with Alexa Fluor 594-conjugated goat anti-rat IgG secondary antibody (1:300, Invitrogen) diluted in PBS. Nuclei were counterstained with DAPI. Finally, fluorescent images were captured using a fluorescence microscope to visualize the labeled proteins and cellular structures.
Statistical Analysis
The data were presented as mean ± standard deviation. Statistical analyses were conducted using SPSS version 16.0 software (SPSS Inc., Chicago, IL, USA). Differences among groups were assessed using either one-way analysis of variance (ANOVA) or Student’s t-test. A P value less than 0.05 was considered statistically significant.




留言 (0)