
記住我
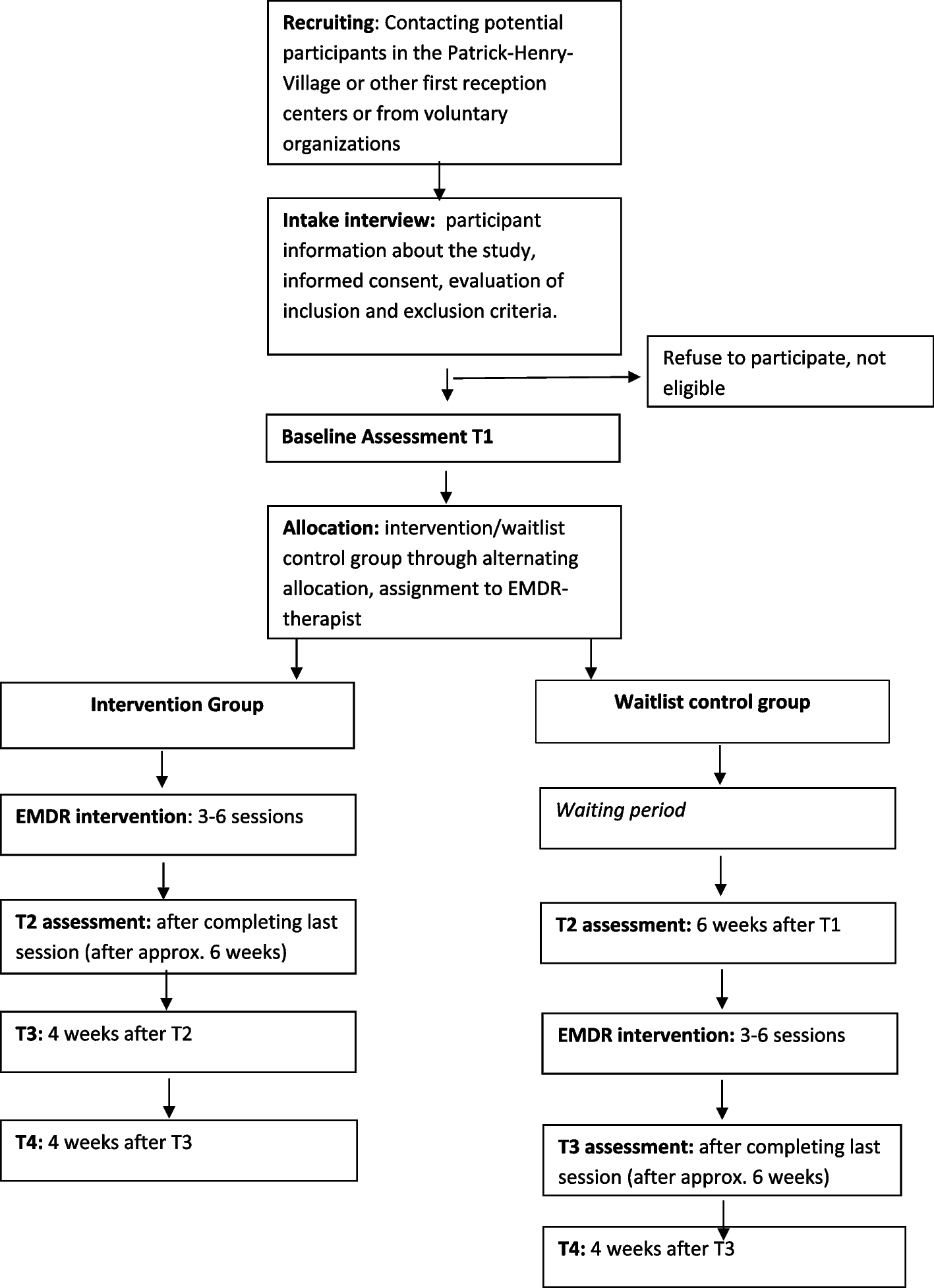


This will be a prospective, parallel-group, single-blind, randomized controlled trial investigating the long-term efficacy and safety of a BDET combined TTI therapy in treating OME, employing pre- and post-operation measurements, with a 24-month follow-up. The protocol was developed following the recommendations of SPIRIT (Standard Protocol Items: Recommendations for Interventional Trials) [16]. A completed SPIRIT checklist was provided as an Additional file.
Trial settingThis investigation will be undertaken within a tertiary care hospital specializing in Ear, Nose, and Throat (ENT), situated in Shanghai. The institution is the largest and most prominent ENT-specialized hospital in China, serving approximately 23 million residents residing in the municipality of Shanghai. It is also a national referral center, extending services to patients from diverse geographical regions across the country.
ParticipantsA total of 124 patients diagnosed as OME at the Eye and ENT Hospital of Fudan University (Shanghai, China) will be included in this trial.
Eligibility screening and enrolmentThe participants’ diagnosis and screening will be administered by two experienced ENT specialists, each with over 10 years of clinical practicing experience. This process will encompass a battery of examinations, including eustachian tube manometry, pure-tone audiometry, ear endoscopy, acoustic impedance measurement, high-resolution CT scans of the temporal bone, and exhaustive medical history assessments. Following these evaluations, eligible participants will be provided with a thorough explanation of the study’s objectives. Should they express their willingness to participate, a face-to-face interview will be scheduled with the patients to answer any questions they might have and ensure they meet all the inclusion and exclusion criteria. Research members from the trial steering committee will introduce the trial to patients during the interview and obtain written consent from patients. The screening procedure will continue until the target population is achieved.
Inclusion criteriaThe inclusion criteria for participants are as follows: (1) a confirmed diagnosis of OME according to the guideline updated in 2016 by the American Academy of Otolaryngology-Head and Neck Surgery Foundation [17], (2) aged > 12 years, (3) presentation of either tympanic membrane retraction or tympanic effusion, as ascertained through meticulous ear endoscopy, (4) CT scan indicating characteristic findings of OME, and (5) confirmation of type B or C tympanograms through the assessment of acoustic impedance.
Exclusion criteriaIndividuals will be excluded if they (1) concurrent manifestation of an upper respiratory tract infection, (2) the presence of documented physiological abnormalities within the ear or nasopharynx, (3) the malformations or aneurysms affecting the ET or the internal carotid artery, as evidenced by high-resolution CT scans, and (5) the incapacity to undergo general anesthesia or adhere to scheduled postoperative follow-up appointments, due to factors unrelated to the study’s objectives.
Withdrawal/retentionParticipation in this study is voluntary, and participants have the right to decide whether to withdraw at any time. We anticipate some dropouts during the follow-up course, considering that the follow-up time is as long as 2 years and there are many kinds of outcome indicators during follow-up. However, we have developed several measures to ensure or improve adherence and to minimize the attrition rate. These include giving participants detailed information from the beginning about the purpose of the study, the treatment options, the importance of follow-up, and the physical benefits they could achieve. Any concerns, such as unexpected symptoms, as well as logistic issues such as traveling to the clinic, parking issues, and making appointments with clinical consultant will be evaluated and solved during each telephone or video consultation. In a limited number of cases, participants may withdraw under certain circumstances, and every reason for withdrawal will be recorded by researchers.
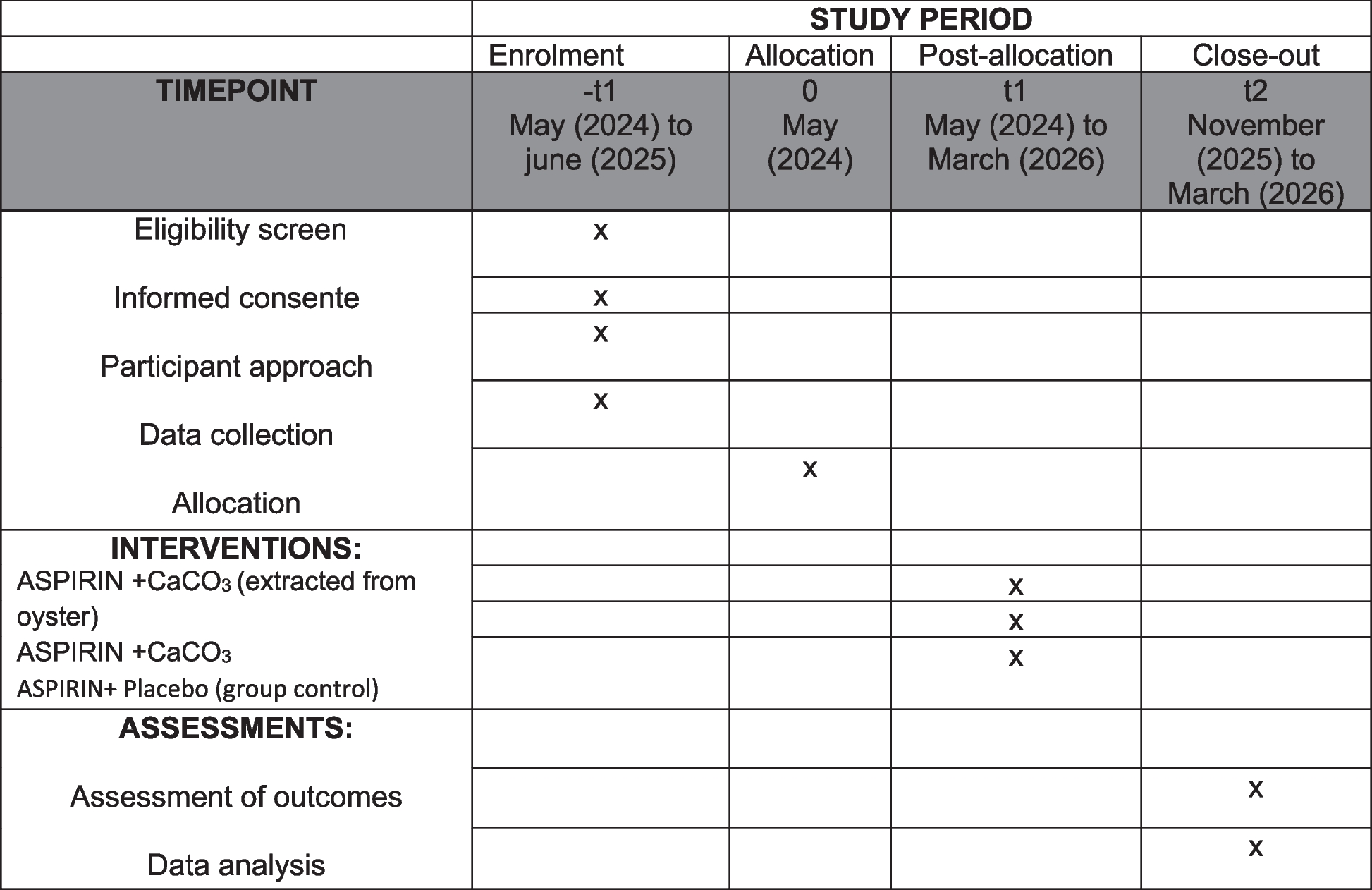
ProcedureUpon obtaining their written informed consent, subjects will then be randomly allocated in a 1:1 ratio to one of two treatment arms: group A, in which participants will undergo conventional therapy with TTI and group B, in which patients will receive BDET combined with TTI therapy. The specific surgical procedure will be elaborated upon in the following section. The same two ENT specialists responsible for the screening procedures will conduct the treatment process for all participants. Furthermore, both groups will concurrently receive the standard level of care and medication within the study’s clinical setting without any restrictions. The assessment of outcomes will take place at five time points, commencing with a baseline evaluation, followed by four post-surgical follow-ups: the first occurring at the 3rd month, the second at the 6th month, the third at the 12th month, and the final at the 24th month. The trial sponsor will bear the additional expenses incurred by patients due to follow-up visits, including transportation costs and examination fees. There is no anticipated harm or compensation for trial participation. A visual representation of the study’s procedural flow can be found in Fig. 1.
Fig. 1 Randomization and blinding
Randomization and blindingParticipants will be randomly assigned to two groups with equal probabilities. Randomization will be conducted by the independent investigator from the trial steering committee, including generating the allocation sequences of the randomization and assigning participants to interventions. The randomization process will be done by an internet-based randomization tool offered by the China Clinical Trial Registration Center, which is freely available at http://www.medresman.org/login.aspx. The randomization sequence will be generated automatically when the participants log into the website and request the allocation number once an eligible patient is recruited. The surgeon will be notified of group allocation after a consent form is obtained and baseline assessments are completed. Considering that BDET is a surgical operation, no blinding will be possible for the surgeon. However, to mitigate potential bias in our trial, patients will be blinded to their treatment assignment, and both data collectors and analysts will maintain blinding to treatment allocation throughout the study. To maintain the overall quality and legitimacy of the clinical trial, unblinding should occur only in exceptional circumstances when knowledge of the actual treatment is absolutely essential for the further management of the patient.
InterventionsFollowing the baseline measurements, participants will be assigned treatments based on randomization outcomes.
Group A will undergo conventional TTI therapy, which will be conducted under general anesthesia and nasal endoscopy. Specialists will select an appropriate size and type of tympanostomy tube based on the patient’s age and middle ear conditions before inserting it into the incision in the eardrum. The procedure concludes with confirming the correct positioning of the tympanostomy tube within the middle ear. Supplementary anti-inflammatory medications may be administered before or after the surgical intervention.
Group B will receive BDET therapy in addition to TTI. After the induction of general anesthesia, patients will be positioned supine with a 30° head elevation. Nasal mucosal vasoconstriction will be achieved using lidocaine and epinephrine cotton tablets on both nasals. A deflated balloon (Manufacturer: Spiggle & Theis Company, Germany) will be introduced into a specialized tube, which will be guided to the pharyngeal opening of the eustachian tube at either a 0° or 30° angle, as visualized through nasal endoscopy. Once the tube aligns parallel to the eustachian tube, the balloon will be carefully advanced into the eustachian cartilage. Subsequently, the balloon will be pressurized to 10 bar for a duration of 2 min. Following this procedure, specialists will release the balloon pressure and remove it from the eustachian tube. Apart from surgical procedures, the care and medication are identical to Group A. A visual representation of the BDET procedure can be found in Fig. 2.
Fig. 2
Balloon dilation eustachian tuboplasty procedure
Outcome measuresPrimary outcomesThe primary outcomes will focus on the eustachian tube function of patients, which will be measured by two tools, both subjectively and objectively:
Eustachian Tube Dysfunction Questionnaire (ETDQ-7)ETDQ-7 serves as an subjective tool for evaluating ET function [18]. It comprises seven items, encompassing ear pain, ear pressure, muffled hearing, ear symptoms during cold or sinusitis, crackling or popping sounds in the ears, tinnitus, and a sense of ear blockage. Each item’s severity is assessed using a 7-point scoring system, ranging from 1 to 7, yielding a maximum total score of 49 points. A higher score indicates more pronounced ear discomfort and greater ET dysfunction.
Eustachian tube score (ETS)The ETS encompasses tubomanometry (TMM) and two subjective questionnaires. TMM serves as an objective tool for assessing the physiological function of the ET [19]. The instrumentation employed for TMM in our study is manufactured by Spiggle & Theis, a German company. The methodology for TMM measurements is consistent with the procedures outlined by Esteve et al. [20], who introduced refinements to the TMM protocol in 2001. During TMM assessments, participants are seated comfortably, and a technician appropriately positions ear probes of suitable dimensions into their external ear canals (EACs), ensuring an airtight seal. Subsequently, participants are instructed to retain a sip of water within their oral cavity. Additionally, a two-pronged nasal probe is snugly inserted into both nostrils, and participants are tasked with maintaining an airtight seal. The TMM apparatus is configured to apply specific pressure values, namely 30 mbar, 40 mbar, and 50 mbar, through the nasal probe situated in the patient’s anterior nostril. Upon sealing the nasal probe valve, participants are directed to swallow. Pressure curves, originating from both the NP and the EACs, are then captured and digitally recorded at these three pressure settings, delineating both temporal (X-axis) and pressure (Y-axis) dimensions. Subsequently, the technician employs a predefined formula to compute the eustachian tube opening latency index, denoted as “R”. Specifically, when 0 < R ≤ 1, it indicates normal ET ventilation function. Conversely, when R > 1, it signifies delayed ET opening, and when R = 0, suggests abnormal ET opening. The absence of an “R” value suggests ET obstruction [21]. To enhance the reliability and objectivity of the data, each TMM test is repeated twice for every patient. An “R” value of ≤ 1 corresponds to a score of 2 points, whereas an “R” value of > 1 is scored as 1 point, and the absence of an “R” value is recorded as 0 points. These scores are then summed across the three distinct pressure levels (30, 40, and 50 mbar, respectively) to yield the objective ETS for the tested ear, which ranges from 0 to 6 points. Subsequently, participants respond to subjective inquiries pertaining to their ability to execute the Valsalva maneuver and their perception of the clicking sound when swallowing. “Always” warrants a score of 2 points, “occasionally” is assigned 1 point, and “never” is indicative of 0 points. These subjective scores are added to the objective ETS score, resulting in a maximum score of 4 points [22]. The cumulative score, inclusive of subjective and objective components, spans a range of 0 to 10 points, with a score of ≤ 5 indicating the presence of Eustachian tube dysfunction.
Secondary outcomesThe secondary outcomes will include middle ear function, hearing situation, and quality of life, which will be measured by:
Acoustic impedance measurements are used to assess the function of the middle ear using the GSI Tympanometer (Tympstar; Grason-Stadler; Inc., Denmark) with a 22 6 Hz 85 dB probe tone. Tympanometric graphs are categorized as type A when peak pressure values fall between − 100 daPa and 50 daPa, and static acoustic admittance values range from 0.3 mL to 1.6 mL. According to the Liden–Jerger classification criteria [23], type A is considered a normal tympanogram, while other types (As, Ad, B, C) are regarded as abnormal tympanograms.
Hearing level, both air conduction (at frequencies of 0.125 kHz, 0.25 kHz, 0.5 kHz, 1 kHz, 2 kHz, 4 kHz, 8 kHz) and bone conduction (at frequencies of 0.25 kHz, 0.5 kHz, 1 kHz, 2 kHz, 4 kHz), will be examined by the GSI audiometer. Pure-tone audiometry will be based on the average hearing thresholds at 0.5 kHz, 1 kHz, and 2 kHz.
CCES will be used to assess the quality of life [24], which comprise three sections: ear symptom perception, social life restrictions, and disease burden, encompassing a total of 13 questions. Each question is assigned a score within a range of 1 to 6 points. Higher scores indicate a lesser degree of disruption caused by the disease, reflecting a better quality of life for those individuals.
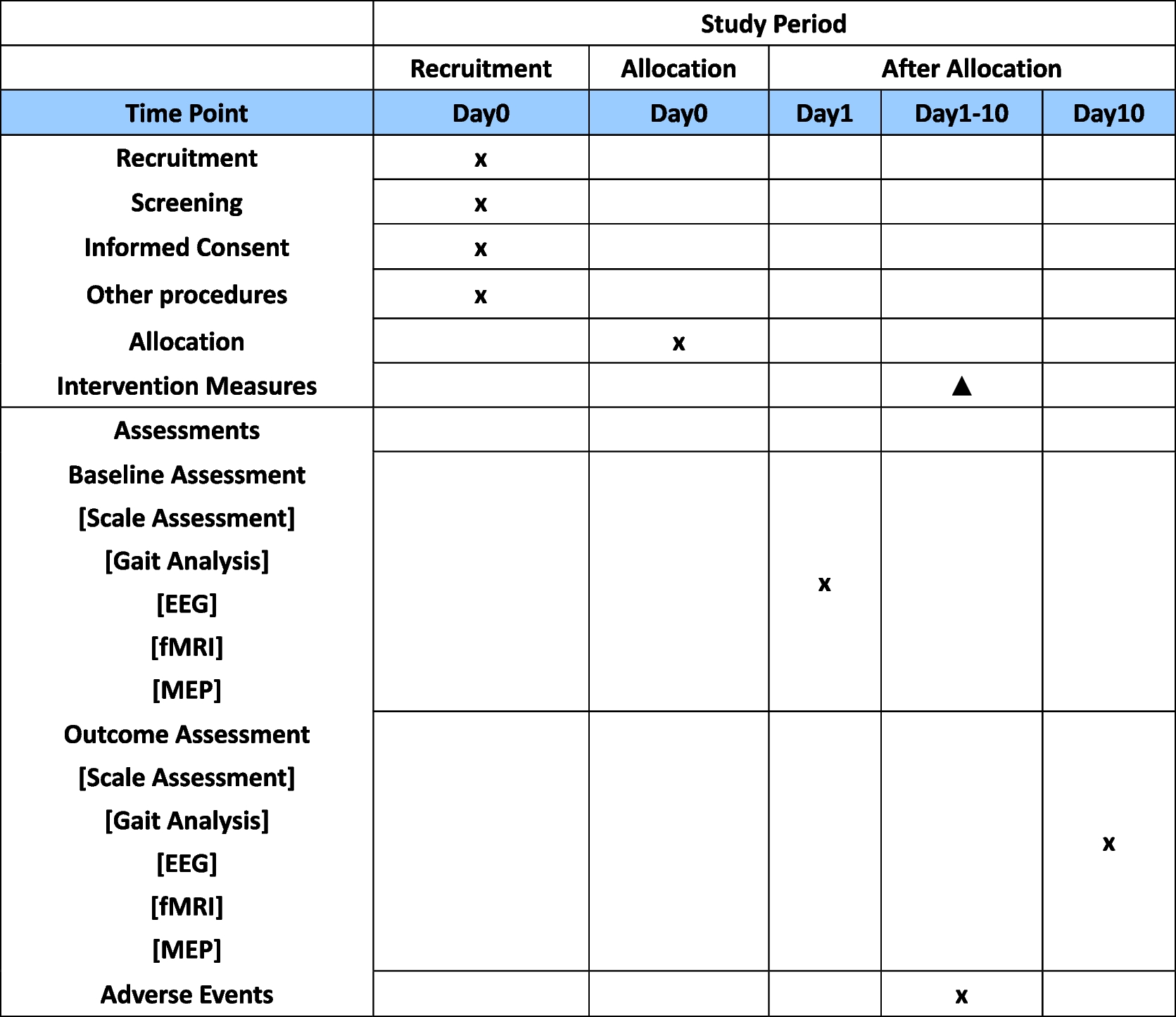
Time-pointsThe assessment of outcomes will take place at the baseline and during four subsequent follow-up visits. Data pertaining to demographics and clinical history will be collected only at the baseline, including age, gender, educational background, employment status, marital status, concurrent systemic ailments, date of initial diagnosis, duration of symptoms, the affected ear, as well as past medication and treatment regimens. Participants will be obliged to return to the clinic at the 3rd, 6th, 12th, and 24th months post-operation. At each follow-up session, they will undergo identical examinations and provide responses to symptom-related questionnaires and tests. A comprehensive schedule outlining the enrollment process, intervention protocols, and the assessment of outcomes is delineated in Table 1.
Table 1 Schedule of enrolment, interventions, and assessmentsSample size calculationThe calculation of the study sample size is based on one of the primary outcomes—EDTQ-7 measurement. The effect size (ES) for the EDTQ-7 was reported to be 1.69 points, with an SD of 2.65, as reported by previous research [25]. Thus, to detect an ES of 1.69 points for the EDTQ-7 with 90% power and an alpha level of 0.05 (two-tailed test), we will require 52 subjects per group. To account for the initially presumed dropout rate of 15%, 62 subjects for each group (124 in total) will be recruited.
Data analysisThe entire statistical analyses will use R software (version 4.2.1 or higher), with a significance level set at p < 0.05. Initial analysis will include summary statistics and visualizations (e.g., histograms, boxplots) to identify outliers and assess data distributions. Subsequently, baseline values will be compared between the two groups. Comparison of adherence and dropout rates will use the chi-squared (χ2) test. Given expected dropouts, the primary analysis will adopt an intention-to-treat (ITT) approach (defined as completing at least one follow-up), with a per-protocol (PP) analysis conducted as secondary. Missing data structure will be examined, with potential application of multiple imputation in ITT analyses.
Primary and secondary outcome changes (e.g., ETS, ETDQ-7, acoustic impedance, PTA, and CCES) will be assessed using mixed-models with repeated measures (MMRM) analyses of variance (ANOVAs). Fixed effects will include group and time, while the subject will serve as a random effect. Models will be adjusted for age, sex, medication history, and other baseline potential confounders.
Statistical analyses will be conducted by an independent, blinded statistician unaffiliated with the study team. There is no planned additional subgroup or adjusted analyses. No interim analyses were planned in this trial.
Adverse eventsMinimal adverse events have been observed in association with balloon dilation eustachian tuboplasty [26]. While the procedure may occasionally lead to nasal and oral mucosal bleeding, the application of epinephrine vasoconstriction can effectively mitigate mucosal congestion and reduce the incidence of bleeding. In cases where bleeding does occur, gentle pressure applied with an epinephrine-soaked cotton pad for a few seconds can effectively staunch the bleeding. Moreover, in this study, participants will be encouraged to promptly report any adverse events following the operation. The research team will maintain regular weekly contact with participants post-surgery to inquire about the emergence of any unexpected symptoms. Any adverse events deemed possibly related to BDET during the trial will be meticulously documented and expeditiously reported to the Adverse Drug Reaction Administration at the Eye and Ear Hospital of Fudan University within a 48-h time frame.
Data managementAll of the data at baseline and the four follow-up visits will be recorded in a self-designed electronic case form. Electronic data will be stored on a public platform at http://www.medresman.org, which is sponsored by the China Clinical Trial Registry. The database uses standard techniques to provide security. Individual participant data (IPD) will be made public 6 months after the completion of the study via the IPD sharing platform.
ConfidentialityTo protect participant anonymity, all subjects will be anonymized, and groups coded as A or B. Encrypted passwords are needed when the researchers or data monitoring group access the database.
Oversight and monitoringComposition of the coordinating center and trial steering committeeThe trial steering committee will comprise one principal investigator and two co-investigators. Their duty is to monitor all aspects of the ongoing progress of the study. Two co-investigators, graduate nursing students, will engage in the patient recruitment process, informed consent, and follow-up. The independent primary investigator will be responsible for generating the allocation sequences of the randomization and assigning participants to interventions. The committee also has the authority to consider and agree upon modifications to the study protocol.
Composition of the data monitoring committee, its role, and reporting structureA data monitoring group will be set up in our research, consisting of a group leader, a statistician, a methodologist, an ENT specialist, and also a patient representative. All members are independent of the study sponsor and report no competing interests.
Frequency and procedures for auditing trial conductThe data monitoring committee will meet regularly on a weekly basis to monitor the data collection and analysis process of the experiment after the trial begins. The trial sponsor will pay for their time and efforts.
Plans for communicating important protocol amendments to relevant partiesAny modifications to the protocol will be reported to the Institutional Review Board of Fudan University Eye Ear Nose and Throat Hospital and also approved by the trial steering committee. Any deviations from the protocol will be fully documented using a breach report.
Dissemination plansWe plan to publish the study findings in peer-reviewed academic journals. We also intend to present this study at local, national, and international conferences where possible. Furthermore, we will draft a summary of the study results to be posted on the website of the Eye and ENT hospital that can be accessed by all trial participants as well as relevant interest groups.
Patient and public involvementPatients and the public were not involved in the design of this study. However, we have consulted with a patient representative about his views on how best to involve patients throughout the proposed project. His views have been incorporated into our revised protocol. Patients and the public will be informed of the study results via peer-reviewed journals or academic conferences.
留言 (0)