
記住我


The trial is designed as a placebo-controlled study with a 1:1 randomization rate between tadalafil and placebo. As we test the feasibility of daily tadalafil treatment in patients with cerebral small vessel disease and its effect on cerebrovascular and cognitive outcomes, placebo treatment was chosen as a comparator.
Intervention descriptionThe intervention is a daily intake of tadalafil 20 mg or placebo together with guideline medication for approximately 3 months. Tadalafil or placebo (lactose monohydrate and potato starch) are included in 96 opaque capsules for each participant to ensure blinding. Participants are instructed to take study medication in the morning. The dose of 20 mg tadalafil was based on our previous study [20], a collaborative study conducted at St George’s University of London, UK [24], and that 20 mg is common in other conditions, such as pulmonary hypertension and erectile dysfunction.
Criteria for discontinuing or modifying allocated interventionsParticipants who repeatedly fail to comply with study agreements can be discontinued. Discontinuation will also occur if the first MRI cannot be conducted as this is considered an important outcome of the trial. In case of serious adverse events due to study medication, the intervention will be stopped. Participants who request to stop study medication, e.g., due to side effects, will be allowed to do so without being discontinued. If study medication is stopped, the participants are encouraged to continue the study without study medication as the feasibility of the treatment is the main outcome. The reason for terminating the study medication is noted.
Strategies to improve adherence to interventionsParticipants are contacted by a phone call every week for the first month after the study medication has been initiated to promote motivation for the study and to assess side effects and adverse events.
Relevant concomitant care permitted or prohibited during the trialApart from nitrates (e.g., isosorbide mononitrate, isosorbide dinitrate, and glyceryl trinitrate) and PDE-5 inhibitors, any treatment or medication is permitted. Nitrates are prohibited due to their interaction with PDE-5 inhibitors. Nitrates and concomitant use of PDE-5 inhibitors during the trial period are listed as exclusion criteria.
Provisions for post-trial careDamages to participants caused by the study are covered, as described in the Danish Law.
OutcomesOutcomes will be analyzed in a main study and the following predefined sub-studies: functional MRI exploratory sub-study, cognitive exploratory sub-study, and biomarker exploratory sub-study. All outcomes in the main study and sub-studies are assessed twice, initially at the baseline visit before study medication is initiated and then at the 3-month follow-up visit at the end of the trial period comparing changes from baseline to follow-up.
Main study Primary outcome: feasibilityThe proportion of participants achieved full target dose of tadalafil/placebo by the end of the trial period. We allow a 10% failure of tablet intake in the trial period which still counts as full completion of medication. Assessment is done by tablet count and a questionnaire.
Secondary outcome: MRICerebral MRI is performed on a 3 T Philips Achieva scanner (Philips, The Netherlands). The following MRI sequences are used for anatomical evaluation: T1w, T2w, fluid-attenuated inversion recovery (FLAIR), diffusion-weighted imaging (DWI), susceptibility-weighted imaging (SWI), and time of flight (TOF). MRIs will be assessed by two independent neuroradiologists according to the STRIVE guidelines from 2013, including white matter hyperintensity (WMH), lacunes, microbleeds, enlarged perivascular space, atrophy, and recent infarcts [25]. We also evaluate the presence of cortical superficial siderosis which is included in the recent STRIVE guidelines from 2023 [26]. WMH volume is assessed using an automatic solution with manual correction based on FLAIR and T1w images [27].
Secondary outcome: cognitionInformant Questionnaire on Cognitive Decline in the Elderly (IQCOED) and Montreal Cognitive Assessment (MoCA) are used to assess cognitive function in the main study.
Secondary outcome: depression, fatigue, and mental well-being questionnairesThe degree of depression is assessed using Becks Depression Inventory II (BDI-II). Fatigue Severity Scale (FSS) is used to examine fatigue, and WHO-5 Well-Being Index to assess mental well-being.
Secondary outcome: safety and adverse eventsAdverse events are registered according to good clinical practice from the day of inclusion until the safety follow-up visit is done.
Secondary outcome: blood pressure, heart rate, and electrocardiogramBlood pressure, heart rate, and electrocardiogram are performed.
Secondary outcomes: blood samplesRoutine blood samples are performed (hemoglobin, hematocrit, leukocytes, platelets, liver-, kidney-, and metabolic function, and high sensitivity C-reactive protein). To control for intake of study medication, plasma levels of tadalafil will be analyzed after the trial is completed.
Secondary outcomes: registered-based follow-upRegister-based outcome assessment will be done with a composite measure of death, any ischemic event, hemorrhagic event, or dementia per patient registry after 3 and 5 years, respectively, from the end of the trial. These outcomes will be published separately.
Functional MRI exploratory sub-studyCalibrated functional MRIWe perform dual-echo pseudo-continuous arterial spin labeling (pCASL) to measure cerebral blood flow (CBF) and blood oxygen level-dependent (BOLD) signals during interleaved normocapnia and hypercapnia (5% CO2, 20% O2 in N2) with/without visual contrast-reversing checkerboard stimulation [28]. From this, we estimate baseline cerebral blood flow (if possible, gray/white-matter perfusion), CVR using hypercapnia, stimulation-induced increase in cerebral metabolic rate of oxygen (CMRO2) and perfusion, and the neurovascular coupling index (NVC) defined as the ratio of those two [29].
BOLD functional MRI (fMRI)We assess changes in the regional event-related BOLD response during a peripheral sensory stimulation of the dominant index finger.
Diffusion-prepared pCASL (DP-pCASL)We perform a sequence set of DP-pCASL scans to map the water exchange across the blood–brain barrier (BBB) [30,31,32,33]. The outcome is a global brain water coefficient (kw).
See Supplementary material 1 for MRI acquisition and stimulation details (Table 3).
Cognitive exploratory sub-studyThe participants undergo a 1.5-h cognitive assessment evaluating the domains of processing speed, verbal and visual working memory, attention, learning and memory, executive functions, and an estimation of premorbid intelligence by the Danish Adult Reading Test (DART). The following paper-and-pencil and Cambridge Neuropsychological Test Automated Battery (CANTAB) tests are done:
Paper-and-pencil: symbol digit modalities test (SDMT), trail making tests A and B, fluency (animal, F-A-S), and digit span forward, backward, ordering, and letter number sequence (from WAIS-IV).
CANTAB: motor screening (MOT), spatial working memory (SMW), paired associates learning task (PAL), reaction time (RTI), one-touch stockings of Cambridge (OTS), and rapid visual information processing (RVP).
Biomarker exploratory sub-studyBlood samples are centrifuged and stored as plasma, serum, and full blood at − 80 °C until the last patient, last visit. Multiplex enzyme-linked immunosorbent assay (Mesoscale, MD, USA) will be used to assess; vascular cell adhesion molecule (VCAM-1), intercellular adhesion molecule-1 (ICAM-1), interleukin-6 (IL-6), tumor necrosis factor alpha (TNF-α), interleukin 1beta (IL-1β), E-selectin, vascular endothelial growth factor, and specific micro-RNAs [20].
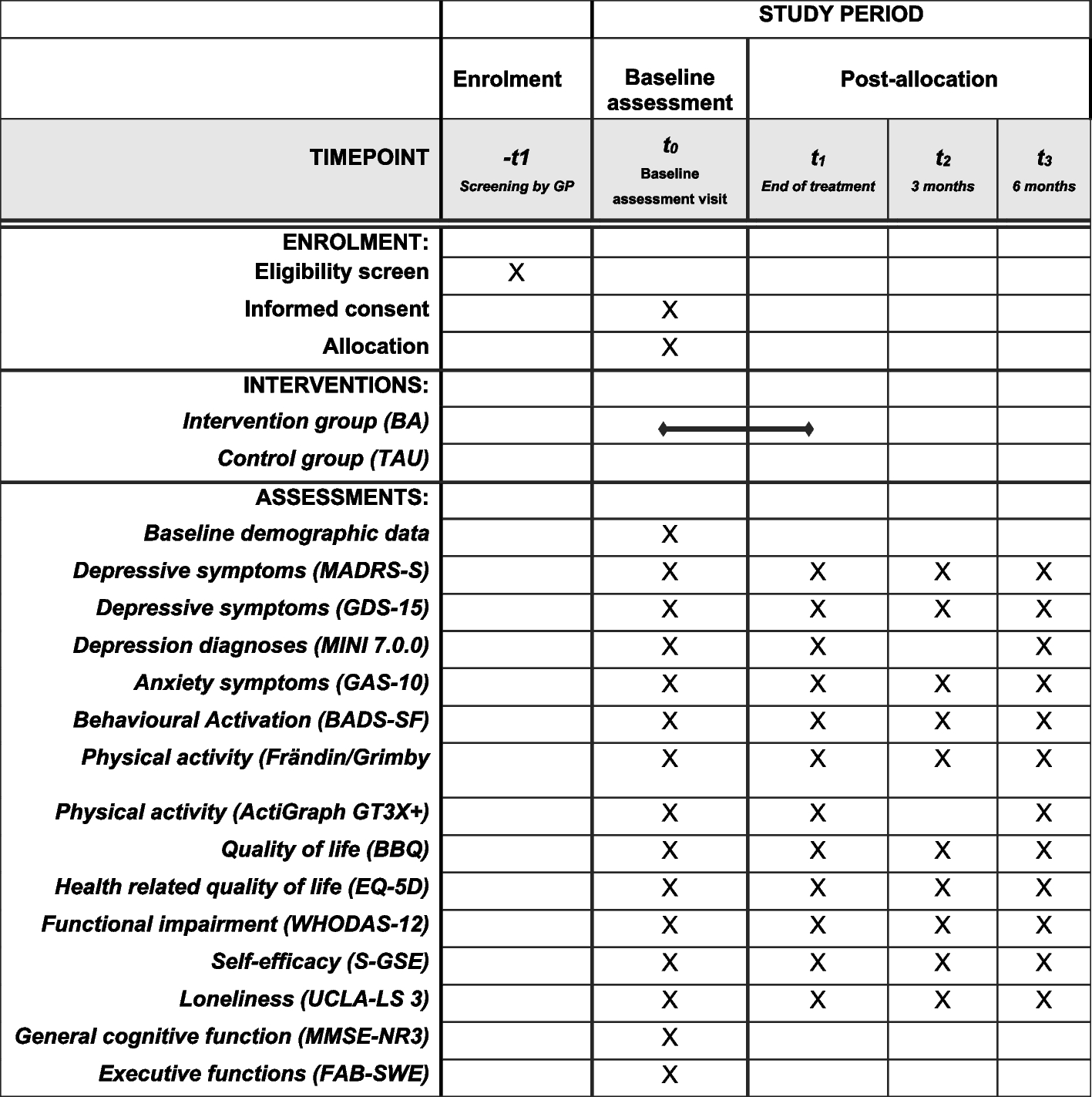
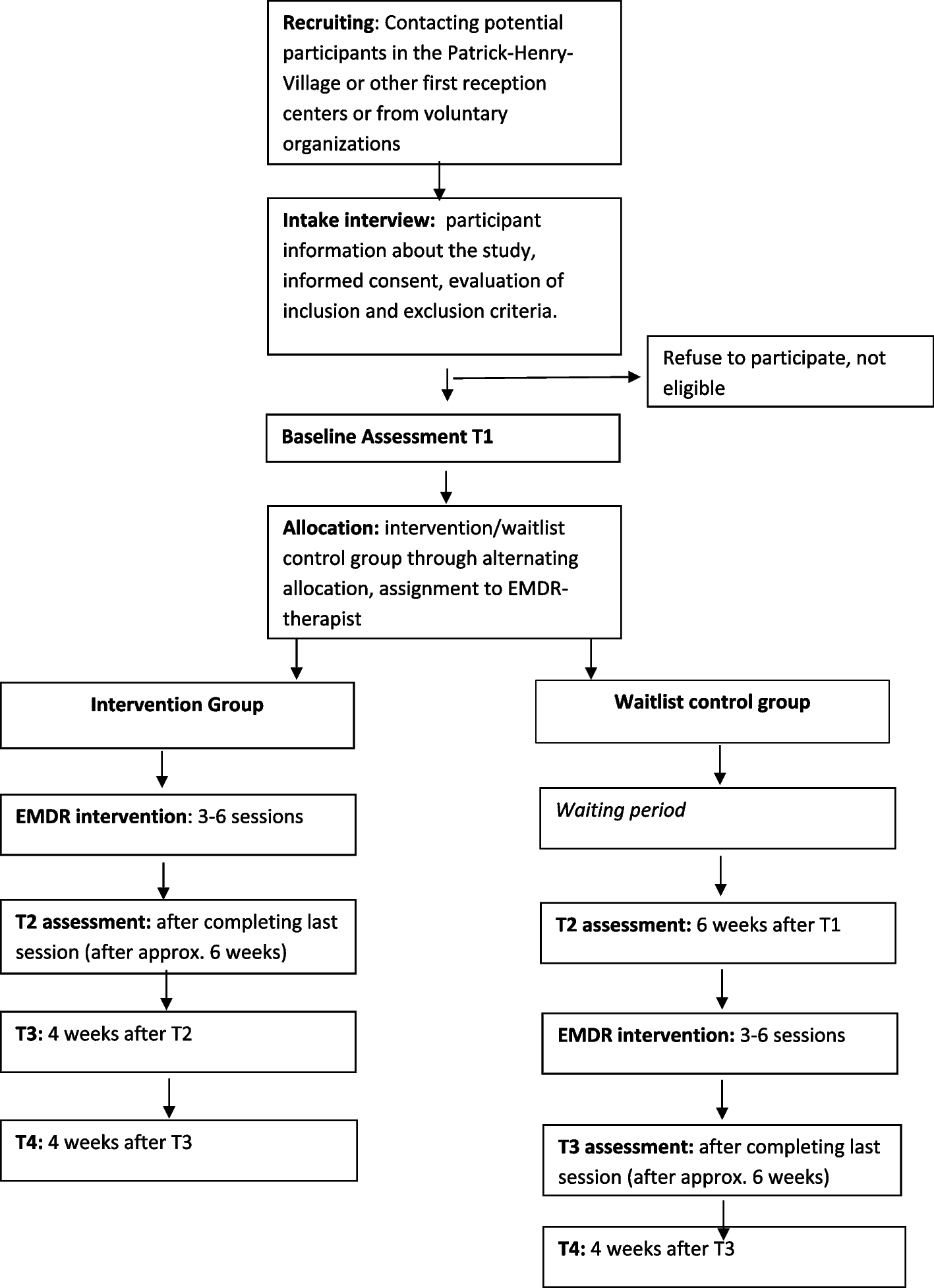
Participant timelineParticipants complete five on-site study visits. Information, inclusion, and the baseline visit can be conducted on the same day. The second visit includes the first MRI examination of the brain (MRI-1), after which the participants can start the study medication. After 1 month of study medication, a follow-up visit is done to assess the side effects. After 3 months of study medication, the second MRI examination (MRI-2) is conducted followed by the 3-month visit after which the study medication is stopped. The participants are contacted by telephone once weekly for the first 3 weeks after the study medication is initiated and after 2 months to assess the side effects. A safety follow-up telephone visit is performed within 2 weeks after the end of the study medication. Visits are performed at HGH and MRIs are done at DRCMR, AHH. See Fig. 1 and Table 4 for visit details.
Fig. 1 Table 4 Schedule and timing of standard protocol assessmentsSample size
Table 4 Schedule and timing of standard protocol assessmentsSample size We hypothesize that 90% of patients in both arms will reach the target dose and complete the trial. By non-inferiority test with a power of 80% and significance level of 0.05, we can detect a difference of 20% between those reaching the target dose on tadalafil compared to placebo with a total sample size of 64 patients. A difference of 20% compared to placebo and 30% in absolute percentage not being able to comply with treatment is considered relevant if to be used in clinical settings. Sample size calculation was also performed for CBF based on 3 T arterial spin labeling (ASL)-MRI for healthy individuals since few 3 T ASL data in patients with CSVD existed when the study was designed. A baseline perfusion of 25 (± 5) ml/100 g/min (mean ± SD) in subcortical white matter, 55 (± 10) ml/100 g/min in cortical gray matter, and 45 (± 10) ml/100 g/min in deep gray nuclei is used for sample size calculations [34,35,36,37,38,39]. To detect a treatment effect of 15% with a statistical power of 80%, a total sample size of 58 is required in subcortical white matter, 50 for cortical gray matter, and 72 for deep gray nuclei. Based on previous studies, we expect a 15% dropout and 10% of patients are estimated not to reach the target dose. To account for a drop-out of up to 25%, we aim to include 100 patients in total. The sample size is calculated using R software (version 3.6.0).
RecruitmentPatients are identified by screening hospital records of previously admitted patients in the neurological departments (HGH, BFH, RH, and NZH). Invitation to participate and trial information is sent to the electronic post-box of eligible patients. The written invitation is followed by a phone call after 1–2 weeks to identify interest in participation. Interested patients are invited to the information and inclusion visit.
留言 (0)