Animals and the PTOA model
Sprague‒Dawley rats (male, 8 weeks of age, n = 54, 220–250 g) were purchased from the Animal Center of Laboratory Animal Center at Beijing Baiaosike Biomedical Technology Co., Ltd. Rats were housed under controlled conditions (25 ± 2 °C, 60% humidity and 12-h light-dark periods). All rats were provided ad libitum access to a standard rodent chow diet and filtered tap water. Rats were allowed to acclimate for 1 week prior to the start of any experiments.
As previously reported [15], anterior cruciate ligament transection (ACLT) was performed on the right knee to cause joint instability, thereby inducing PTOA. The rats were randomized into the following four groups (n = 6 rats/group): a sham group, an ACLT group, an ACLT + vector, and an ACLT + pcDNA-NUMB group. In brief, rats in the final three groups were anesthetized via intraperitoneal injection of sodium pentobarbital. After disinfection, the medial skin of the patellar ligament was incised, and the joint capsule was opened to dislocate the patellar bone. The knee joint was flexed to expose the anterior cruciate ligament, and the anterior cruciate ligament was cut. After rinsing with sterile saline, the wound was closed layer by layer and disinfected, and penicillin was injected intramuscularly to prevent infection. After 6 weeks, the rats were anesthetized with xylazine (5 mg/kg) and ketamine (100 mg/kg), sacrificed by asphyxiation with CO2 and the knee joints were harvested. All surface soft tissues (skin, muscle, etc.) were removed, and knee joints were fixed in 4% buffered formalin for 48 h at room temperature. Fixed rat knees were decalcified with 10% EDTA solution for 30 days, and the EDTA solution was replaced every 3 days. Then, these specimens were embedded in paraffin and serially cut into 3-µm thick sections for subsequent staining of pathological sections. Besides, the cartilage tissue and blood were immediately removed, t and the latter was collected to prepare the serum for subsequent experiments. In the sham control mice, a skin and capsule incision was instead made. Animal experiments were approved and supervised by the Animal Ethics Committee of No. 971 Hospital of the PLA Navy. All methods were carried out in accordance with relevant guidelines and regulations.
Safranin O/Fast green staining and OARSI scoring
Cartilage tissue sections (n = 6 rats/group) were dewaxed with 4% xylene and dehydrated with gradient alcohol. The tissue sections were stained with Fast Green for 10 min and washed with water until the cartilage appeared colorless and was differentiated in acidic ethanol differentiation solution. Then, Safranin O solution (ScyTek, Logan, UT, USA) was used to stain the tissue sections for 30 s, followed by xylene transparency for 5 min and mounting on neutral resin. The presence of any morphological changes in the tissue sections was evaluated in a blinded manner by three certified pathologists using a BX51 optical microscope (Olympus Corp., Tokyo, Japan).
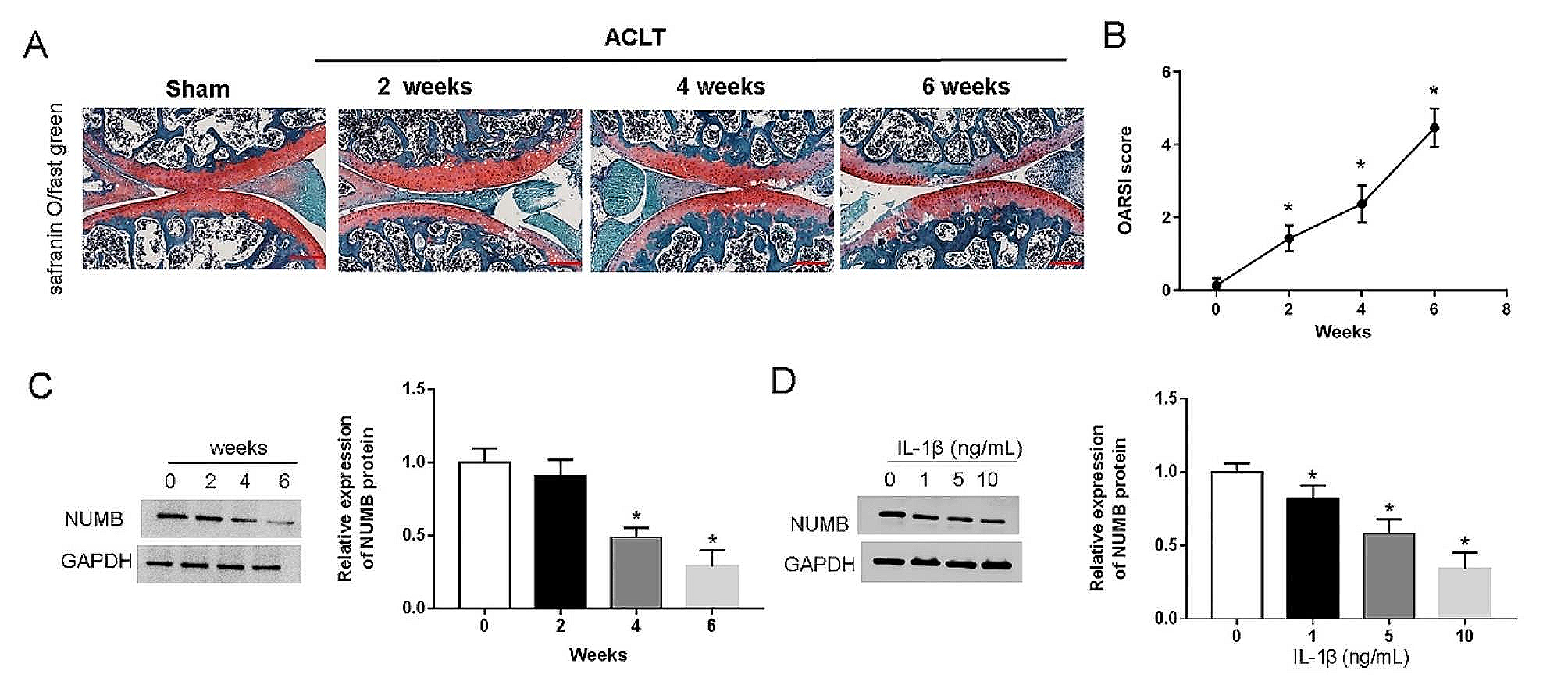
Rat cartilage degeneration in the stained sections was graded using the Osteoarthritis Research Society International (OARSI) scoring system. Specifically, a subjective score of 0–6 was applied as previously described [16]. Scoring criteria were as follows: 0 = normal cartilage; 0.5 = loss of Safranin-O without structural changes; 1 = a small amount of fibrosis but no cartilage loss; 2 = surface fissures with a small amount of cartilage loss, 3 = fissures and erosions are deep to the calcified cartilage layer and cover less than 25% of the articular surface; 4 = fissures and erosions are deep to the calcified cartilage layer and over an area of 25–50% of the articular surface; 5 = fissures and erosions are deep to the calcified cartilage layer and over an area of 50–75% of the articular surface; and 6 = fissures and erosions are deep to the calcified cartilage layer and cover more than 75% of the articular surface. Three blinded observers graded each section. The three grades for each section were then averaged, and the data for the rat in each group were collated. Higher scores were indicative of more serious cartilage damage.
Hematoxylin-eosin (HE) staining
Cartilage tissue sections (n = 6 rats/group) were stained with hematoxylin-eosin (H&E) solution. The percentage of staining-positive area was then determined by pathologists using a BX51 microscope at a magnification of ×400 to evaluate the degree of cartilage injury.
Cell culture
Human primary chondrocytes were purchased from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). The cells were cultured in complete Dulbecco’s modified Eagle’s medium (DMEM; Gibco, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (FBS; Gibco, Grand Island, NY, USA), streptomycin (100 mg/mL) and penicillin (100 U/mL) (Sigma, St. Louis, MO, USA) at 37 °C in a humidified atmosphere containing 5% CO2. Only cells within the fifth passage were used for the subsequent experiments.
Cell treatment
Chondrocytes were harvested as above, counted, and seeded into 96-well plates at a density of 104 cells/well (0.1 mL/well). At 24 h postseeding, the cells were treated with various doses of IL-1β (1, 5, or 10 ng/mL; Sigma Aldrich, St. Louis, MO, USA) as a cell inflammatory model, and normal chondrocytes treated with PBS were used as a control.
The short-hairpin NUMB (sh-NUMB) plasmid, short-hairpin BTRC (sh-BTRC) plasmid, and their corresponding negative control (sh-NC), as well as the NUMB and BTRC overexpression plasmids (pcDNA-NUMB and pcDNA-BTRC) and their corresponding negative control (vector), were synthesized by GenePharma (Shanghai, China). Chondrocytes were grown to 70-80% confluence in 24-well plates and then transfected with the above plasmids with Lipofectamine 3,000 reagent (Invitrogen, CA, USA) according to the manufacturer’s instructions. After transfection for 48 h, the chondrocytes were collected for subsequent experiments.
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay
Cell viability was evaluated using an MTT assay. In brief, after the indicated transfection or treatment, the chondrocytes were harvested, washed with PBS, and counted, after which 1 × 104 chondrocytes were subsequently seeded into 96-well plates. The plates were then incubated at 37 °C for 72 h before each well was treated with 20 µL of 0.5 mg/mL MTT (Beyotime, Shanghai, China) for 4 h. After the supernatant was removed, 150 µL of dimethyl sulfoxide (Sigma Aldrich, St. Louis, MO, USA) was added to each well (Sigma Aldrich, St. Louis, USA). After that, the optical density (OD) was subsequently evaluated at 490 nm using an automated microplate reader (Molecular Devices, San Jose, CA) to determine the number of viable cells.
TdT-mediated dUTP Nick-End labeling (TUNEL) staining
After transfection, TUNEL staining was performed using a TUNEL fluorescence kit (Roche, Basel, Switzerland) to detect apoptosis in chondrocytes in the presence of IL-1β (10 ng/mL). In brief, the chondrocytes were harvested, rinsed with PBS, counted, and seeded onto glass coverslips at 103 cells/coverslip. The cells were then fixed by coating each coverslip with acetone/methanol (vol/vol) for 5 min and placing them at -20 °C. Then, the chondrocytes were incubated with equilibration buffer for 10 s before terminal deoxynucleotidyl transferase (TdT) was added for 1 h at 37 °C. Subsequently, the chondrocytes were incubated with the reaction mixture for 60 min, and the cell nuclei were stained with DAPI. The images/slides were observed by using an inverted fluorescence microscope (Nikon, Tokyo, Japan). A visiopharm image analysis software (Version 6.7.0.2590; Visiopharm, Westminister, CO, USA) was used to count TUNEL-positive stained cells. The number of TUNEL-positive apoptotic chondrocytes are presented as (TUNEL-positive cells)/(total cells)×100%.
Western blotting
Cartilage tissues (50 mg/rat) and transfected chondrocytes were collected and lysed with RIPA buffer containing protease inhibitors (Beyotime, Jiangsu, China). Following the quantitation of protein content using a bicinchoninic acid (BCA) protein assay kit (Beyotime, Shanghai, China), equal amounts of protein (20 µg) were loaded into the wells of a 10% SDS‒PAGE gel, and the proteins were resolved. Thereafter, the proteins were electrotransferred to polyvinylidene difluoride (PVDF) membranes (Millipore, Billerica, MA). For each protein of interest, a dedicated blot was generated (this would reduce the risk of antigen loss due to stripping/reprobing one membrane over and over). Nonspecific binding on each membrane was blocked by coating the membrane with a solution of 10 mM Tris-buffered saline (TBS) containing 5% nonfat milk and then incubating the mixture at 25 °C for 2 h. The membranes were incubated with primary antibodies purchased from Abcam (Cambridge, UK), and the antigens used were NUMB (1: 1,000), BTRC (1: 1,000), p-p65 (1:500), p65 (1: 1,000), IκBα (1: 1,000), and p-IκBα (1: 1,000). In all the cases, an antibody against GAPDH (1: 1,000) was also included (as a housekeeping/protein loading protein). All the membranes were then incubated at 4 °C for at least 12 h. Thereafter, each membrane was gently rinsed with TBS containing Tween-20 (TBST), coated with TBST containing horseradish peroxidase-conjugated secondary antibody (1: 5,000) and incubated at room temperature in the dark for another 2 h. Immunoblots were visualized by an enhanced chemiluminescence detection kit (ECL kit; Millipore, Billerica, MA) under a chemiluminescence imaging analysis system (Amersham Imager 600, GE, CT, USA). Relative integrated density values were calculated using ImageJ software provided by the National Institutes of Health (Bethesda, MD, USA).
Immunofluorescence staining
Chondrocytes were inoculated on polylysine-coated cover glass for 24 h, fixed with 4% paraformaldehyde for 20 min, and permeated with PBS containing 0.2% Triton X-100 for 10 min. Then, 5% BSA in PBS was added dropwise to the slides, which were blocked for 30 min at room temperature. Thereafter, the slides were coated with a solution of PBS containing a 1:200 dilution of the primary antibody against NF-κB p65 (Abcam, Cambridge, UK) at 4 °C overnight. The cells were incubated with a PBS solution containing a 1:500 dilution of Alexa Fluor® 555-conjugated secondary goat anti-rabbit antibodies (Cell Signaling Technology, Danvers, USA). The samples were incubated in the dark for 1 h at room temperature before they were gently rinsed with PBS, after which the cells were counterstained with DAPI solution (5 µg/mL) (Beyotime, China) for 10 min. Images were observed by an Olympus fluorescence microscope (Nikon, Tokyo, Japan) to determine the fluorescence intensity with Image-Pro Plus 6.0 (NIH, Bethesda, MD, USA).
Enzyme-linked immunosorbent assay (ELISA)
The levels of TNF-α and IL-6 in the serum and in the supernatants from cultured chondrocytes were measured with commercial ELISA kits (R&D Systems, MN, USA) following the manufacturer’s instructions. Similarly, the levels of related secretory proteins, such as MMP-13 and COL2A1, were also detected via ELISA kits (R&D Systems, MN, USA). The limits of detection of the kits were 7.2 pg/TNF-α/mL, 1.8 pg IL-6/mL, and 21.3 pg MMP-13/mL. The level of COL2A1 was detected by sandwich ELISA and an anti-COL2A1 antibody (1: 1,000). All the samples were evaluated in triplicate.
Coimmunoprecipitation (Co-IP) assay
Chondrocytes (1 × 107 total/treatment) were lysed in RIPA buffer (Beyotime, Shanghai, China) following the manufacturer’s protocols. After centrifugation at 12,000 rpm for 10 min, the resulting cell supernatant was collected, aliquoted (500 µL), combined with a PBS solution containing mouse anti-human NUMB antibody (1 : 30), anti-human BTRC antibody (1 : 30), or nonspecific IgG (1 : 50) and incubated overnight at 4 °C. Thereafter, the mixture was treated with 50 µL of a kit-provided solution containing protein A/G agarose beads (Takara Biotechnology, Dalian, China), and the sample was then incubated overnight at 4 °C. After this second incubation, the mixture was centrifuged at 3000 rpm for 5 min, and the beads were collected and washed with phosphate-buffered saline (PBS, pH 7.4) three times. The beads were then boiled in loading buffer for 5 min and centrifuged (3000 rpm, 5 min) to allow the adherent proteins to separate from the beads. All precipitated (IP) products were isolated in this manner and subsequently analyzed via Western blotting.
Statistical analysis
All the statistical analyses were performed with SPSS software (version 28.0, IBM Corp., Chicago, IL, USA), and the results are presented as the means ± standard deviations (SDs). Student’s t test was used for comparisons between two groups, and one-way analysis of variance (ANOVA) was used for comparisons among groups. P < 0.05 was considered to indicate a statistically significant difference.



留言 (0)