
記住我



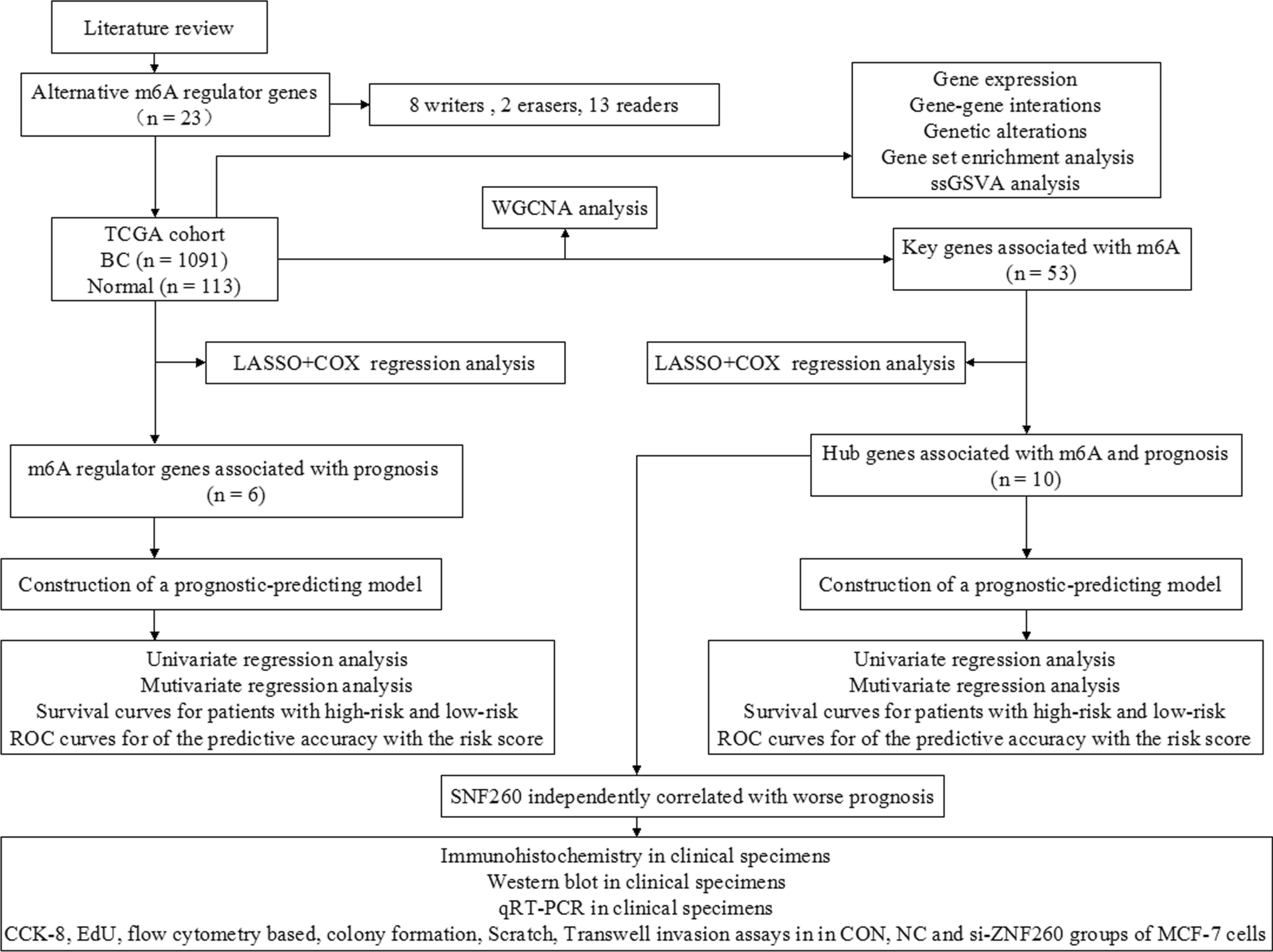
Our workflow is shown in Fig. 1. The GSE112790 dataset included 183 HCC tumors and 15 adjacent liver samples. The differentially expressed genes (DEGs) were screened with the following criteria: |logFC|> 1 and adj P < 0.05. As shown in Fig. 2A, these autophagy-related genes could distinguish between paracarcinoma tissue samples and HCC tissue samples, suggesting that autophagy-related genes play essential roles in the development of HCC. A total of 1408 differentially expressed mRNAs (DEMs) were identified between cancer tissue samples and paracarcinoma tissue samples in the dataset (Fig. 2B). Venn diagrams were drawn using an online Venn diagrams tool. There were 57 autophagy-related DEGs (Fig. 2C), including 25 upregulated DEGs (Fig. 2D, Table 1) and 32 downregulated DEGs (Fig. 2E, Table 1).
Fig. 1 Fig. 2
Fig. 2
Identification of autophagy-related genes using GEO and TCGA datasets. A PCA array. B DEGs between the 183 HCC tumors and 15 adjacent liver samples. C Heatmap showing the expression levels of 57 autophagy-related DEGs in normal and tumor tissues. D Twenty-five overlapping genes identified as autophagy-related upregulated DEGs. E Thirty-two overlapping genes identified as autophagy-related downregulated DEGs
Table 1 Lists of DEGs upregulated/downregulated in dataset GSE1127903.2 Enrichment analysis of autophagy-related genesIn this study, we performed gene set enrichment analysis to observe the functional enrichment of autophagy-related DEGs in HCC (Table 1).
Gene Ontology (GO) enrichment analyses were conducted for the upregulated (Table 2) and downregulated DEGs related to autophagy (Table 3).
Table 2 GO-BP and KEGG pathway analyses of upregulated DEGs associated with autophagyTable 3 GO-BP enrichment and KEGG pathway analyses of downregulated DEGs associated with autophagyThe top 10 pathways identified by the GO analyses are displayed in a bubble diagram.
The coupregulated DEGs were mostly enriched in protein catabolic process, macroautophagy, peptidyl serine phosphorylation, peptidyl-serine modification and regulation of cellular protein catabolism process (Fig. 3A), and the codownregulated DEGs were mostly enriched in regeneration, response to extracellular stimuli, epithelial cell proliferation, response to nutrient levels, and positive regulation of cellular catabolic processes and regeneration (Fig. 3C).
Fig. 3
Enrichment analysis of autophagy-related genes by TCGA database. A GO-BP enrichment analysis of autophagy-related upregulated genes. B KEGG enrichment analysis of autophagy-related upregulated genes. C GO-BP enrichment analysis of autophagy-related downregulated genes. D KEGG enrichment analysis of autophagy-related downregulated genes
Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses were conducted for the upregulated (Table 2) and downregulated DEGs related to autophagy (Table 3).
The top 10 pathways identified by the KEGG analyses are displayed in a bubble diagram.
The coupregulated DEGs were mostly enriched in fluid shear stress and atherosclerosis, focal adhesion and the PI3K-Akt signaling pathway (Fig. 3B), and the codownregulated DEGs were mostly enriched in autophagy–animal and cytokine‒cytokine receptor interactions (Fig. 3D).
3.3 Construction of a protein‒protein interaction (PPI) network via STRING and identification of HUB genes via Cytoscape softwareSTRING was used to construct a network for the DEGs associated with autophagy (Fig. 4A). After further analysis with Cytoscape, IGF1, CDKN2A, BIRC5, and SPP1 were selected as the HUB genes and visualized (Fig. 4B). IGF1, CDKN2A, BIRC5, and SPP1 were selected as potential biomarkers (Table 4) and further validated in subsequent studies.
Fig. 4
Identification of HUB genes by the STRING database. A PPI network of overlapping DEGs. B The most significant 15 node degree genes calculated by the cytoHubba app in Cytoscape. IGF1, CDKN2A, BIRC5, and SPP1 were selected as the HUB genes. The node color intensities representing different genes correlate with the degree of expression values
Table 4 Information on differentially regulated genes3.4 Database-based validation of the transcript levels of IGF1, CDKN2A, BIRC5, and SPP1The expression levels of IGF1, CDKN2A, BIRC5, and SPP1 mRNAs in paired (Fig. 5A–D) and nonpaired (Fig. 5E–H) samples in the TCGA database were evaluated. CDKN2A, BIRC5, and SPP1 expression levels were significantly upregulated in tumor tissues (p < 0.001) (Fig. 5A–H), while IGF1 expression was considerably downregulated (p < 0.001). We also used a GSE84402 dataset to validate the mRNA expression levels of IGF1, CDKN2A, BIRC5, and SPP1 in hepatocellular carcinoma tissues and adjacent tissues. Compared with those in adjacent tissues, the expression levels of CDKN2A, BIRC5, and SPP1 were significantly upregulated in tumor tissues (p < 0.05), while the expression of IGF1 was considerably downregulated (p < 0.001) (Fig. 5I–L). Furthermore, immunohistochemical staining images from the HPA database indicated that the expression levels of CDKN2A, BIRC5, and SPP1 were upregulated in tumor samples, while IGF1 expression was not detected in either hepatocellular carcinoma tissues or adjacent tissues (Fig. 6). This finding was different from our previous analysis. We suspected that the author for the dataset did not choose an effective antibody or that the experimental conditions needed to be explored. These findings indicated that CDKN2A, BIRC5, and SPP1 were upregulated in HCC, while IGF1 expression was markedly downregulated.
Fig. 5
Expression levels of IGF1, CDKN2A, BIRC 5, and SPP1 in HCC from the GEO and TCGA databases. A–H IGF1, CDKN2A, BIRC5, and SPP1 mRNA expression levels are based on the TCGA database, including paired (A–D) and nonpaired samples (E–H). I–L The mRNA expression levels of IGF1, CDKN2A, BIRC5, and SPP1 mRNA are based on GSE84402
Fig. 6
Levels of IGF1, CDKN2A, BIRC5, and SPP1 in normal individuals and patients with HCC from the HPA database
3.5 Diagnostic value of IGF1, CDKN2A, BIRC5 and SPP1 for HCCThis study investigated the clinical diagnostic value of IGF1, CDKN2A, BIRC5, and SPP1 in HCC by constructing ROC curves. The areas under the curve (AUCs) were 0.731, 0.953, 0.982, and 0.734, indicating the diagnostic value of IGF1, CDKN2A, BIRC5, and SPP1 for HCC, respectively (Fig. 7A–D).
Fig. 7
Analysis of the clinical implications of IGF1, CDKN2A, BIRC5, and SPP1 on the XIANTAO planform. A–D ROC curve analysis for IGF1, CDKN2A, BIRC5, and SPP1. E–H Overall survival analysis for IGF1, CDKN2A, BIRC5, and SPP1. I–L Analysis of the association of TNM stages with IGF1, CDKN2A, BIRC5, and SPP1
3.6 Overall survival analysis for patients with HCC stratified by IGF1, CDKN2A, BIRC5 and SPP1 levelsThe overall survival of patients with HCC stratified by IGF1, CDKN2A, BIRC5, and SPP1 levels was analyzed by Kaplan‒Meier plotter to further investigate whether IGF1, CDKN2A, BIRC5, or SPP1 had an effect on overall survival. Our findings suggested that high levels of CDKN2A, BIRC5, and SPP1 were associated with poor overall survival in patients with HCC, suggesting that BIRC5, CDKN2A, and SPP1 are associated with HCC progression and could be used as tumor biomarkers in patients with HCC. IGF1 was at lower levels in tumor tissues than in adjacent tissues, possibly because of its association with lower survival (Fig. 7E–H).
3.7 Analysis of the associations of tumor node metastasis (TNM) stages with IGF1, CDKN2A, BIRC5 and SPP1 levelsWe analyzed the associations of TNM stage with IGF1, CDKN2A, BIRC5, and SPP1. We found significant differences in different T stages compared to regular level, suggesting that HUB genes may be related to the progression of HCC (Fig. 7I–L).
3.8 GSEA revealed pathways associated with IGF1, CDKN2A, BIRC5 and SPP1 expression in HCCGSEA indicated that IGF1-related genes were mainly enriched in immune-related pathways, such as disease of the immune system, immunoregulatory interactions between lymphoid and nonlymphoid cells, drug metabolism, other enzymes, and drug ADME (Fig. 8A). CDKN2A-related genes were mainly enriched in the mitotic G1 phase and G1 transition, integrated cancer pathway, regulation of TP53 activity, and disease of programmed cell death (Fig. 8B). BIRC5-related genes were mainly enriched in the mitotic G1 phase and G1 transition, immunoregulatory interaction, lymphoid and nonlymphoid cells, retinoblastoma gene in cancer, and FceRI-mediated NFkb activation (Fig. 8C). SPP1-related genes were mainly enriched in pathways related to cancer, immunoregulatory interactions between lymphoid and nonlymphoid cells, photodynamic therapy-induced NFkb survival signaling, focal adhesion PI3K, AKT, and the mTOR signaling pathway (Fig. 8D).
Fig. 8
GSEA for IGF1, CDKN2A, BIRC5, and SPP1 by the TCGA database
3.9 Correlation analysis of IGF1, CDKN2A, BIRC5 and SPP1 with the TME in HCCAs demonstrated in Fig. 9A, patients with lower level of IGF1 had lower numbers of memory dendritic cells (DCs), aDCs, cytotoxic cells, mast cells, neutrophils, Tgd, Th1 cells, Th17 cells, and Tregs. Additionally, as depicted in Fig. 9B–D, patients in the groups with high CDKN2A, BIRC5, and SPP1 had high Th2 cells and low Th 17 cells and eosinophils. According to Fig. 9E, patients with high IGF1 had negative correlations with NK and CD56bright cells but positive correlations with neutrophils, DCs, cytotoxic cells, Tregs, Th1 cells, Th17 cells, Mast cells, Tgd and NK CD56dim cells. As demonstrated in Fig. 9F–H, patients with high CDKN2A, BIRC5 and SPP1 had negative correlations with Th17 cells and eosinophils but positive correlations with Th2 cells. Neutrophils, Th17 cells, and eosinophils may be the most prevalent differential immune cells. Th2 cells may play a role in promoting cancer. In this research, we further assessed the correlations between immune infiltration levels and the expression levels of IGF1, CDKN2A, BIRC5, and SPP1 in HCC via the TIMER database. As shown in Fig. 10, IGF1 was negatively correlated with the infiltration of macrophages (p = 1.13 × 10 − 2); moreover, CDKN2A, BIRC5, and SPP1 levels were positively correlated with the infiltration of macrophages, B cells, CD4 + T cells, CD8 + T cells, dendritic cells, and neutrophils. Figure 11A–D shows that there was variability in the stromal, immune, and estimated scores of patients with HCC, indicating some differences in the TME according to the level of IGF1, CDKN2A, BIRC5, and SPP1. Various immune checkpoints were associated with IGF1, CDKN2A, BIRC5, and SPP1 (Fig. 11E).
Fig. 9
Levels of IGF1, CDKN2A, BIRC5, SPP1 and their correlations with the TME on the XIANTAO planform. A–D Immune infiltration analysis for IGF1, CDKN2A, BIRC5 and SPP1. E–H Correlation levels of IGF1, CDKN2A, BIRC5, and SPP1 with the level of immune infiltration
Fig. 10
Immune infiltration analysis of IGF1, CDKN2A, BIRC5, and SPP1 performed using the TIMER database
Fig. 11
Immune analysis of IGF1, CDKN2A, BIRC5, and SPP1 by the TCGA database. A–D Correlations of the estimated proportions of immune and stromal cells with IGF1, CDKN2A, BIRC5, and SPP1 levels in HCC. E Various immune checkpoints were associated with IGF1, CDKN2A, BIRC5, and SPP1
3.10 Possible sensitivity to therapeutic drugsUsing the CADSP database, we screened 288 drugs and identified antitumor drugs that were relatively sensitive to DEGs. Patients with high level of CDKN2A and BIRC5 had the lowest half maximal inhibitory concentrations (IC50) for Epothilone B, while SPP1 was inhibited by bortezomib. Shikonin (SHK) had the lowest IC50 in patients in the group with high IGF1 level. We identified 35 tumor-sensitive drugs targeting the HUB genes (Fig. 12).
Fig. 12
Possible Sensitivity to Therapeutic Drugs according to the CADSP database
3.11 External validation in patients with HCC from the hospitalWe validated the protein levels of IGF1, CDKN2A, BIRC5, and SPP1 in HCC by IHC (Fig. 13). CDKN2A, BIRC5, and SPP1 were upregulated in HCC tissues by IHC. However, IGF1 was downregulated in HCC tissues compared with adjacent tissues.
Fig. 13
Levels of IGF1, CDKN2A, BIRC5, and SPP1 in patient tissues by IHC. Levels of CDKN2A, BIRC5, and SPP1 in HCC were increased compared with tumor-adjacent tissues. However, IGF1 expression was downregulated in HCC tissues compared with adjacent tissues
We also validated the expressions of IGF1, CDKN2A, BIRC5, and SPP1 in HCC by RT‒PCR (Fig. 14). CDKN2A, SPP1, and BIRC5 were upregulated in HCC tissues according to RT‒PCR; IGF1 was downregulated in HCC tissues compared with adjacent tissues.
Fig. 14
Expressions of IGF1, CDKN2A, BIRC5, and SPP1 in patient tissues by RT‒PCR. The expressions of CDKN2A, SPP1, and BIRC5 in HCC were increased compared with tumor-adjacent tissues. However, IGF1 expression was downregulated in HCC tissues compared with that in adjacent tissues
留言 (0)