
記住我
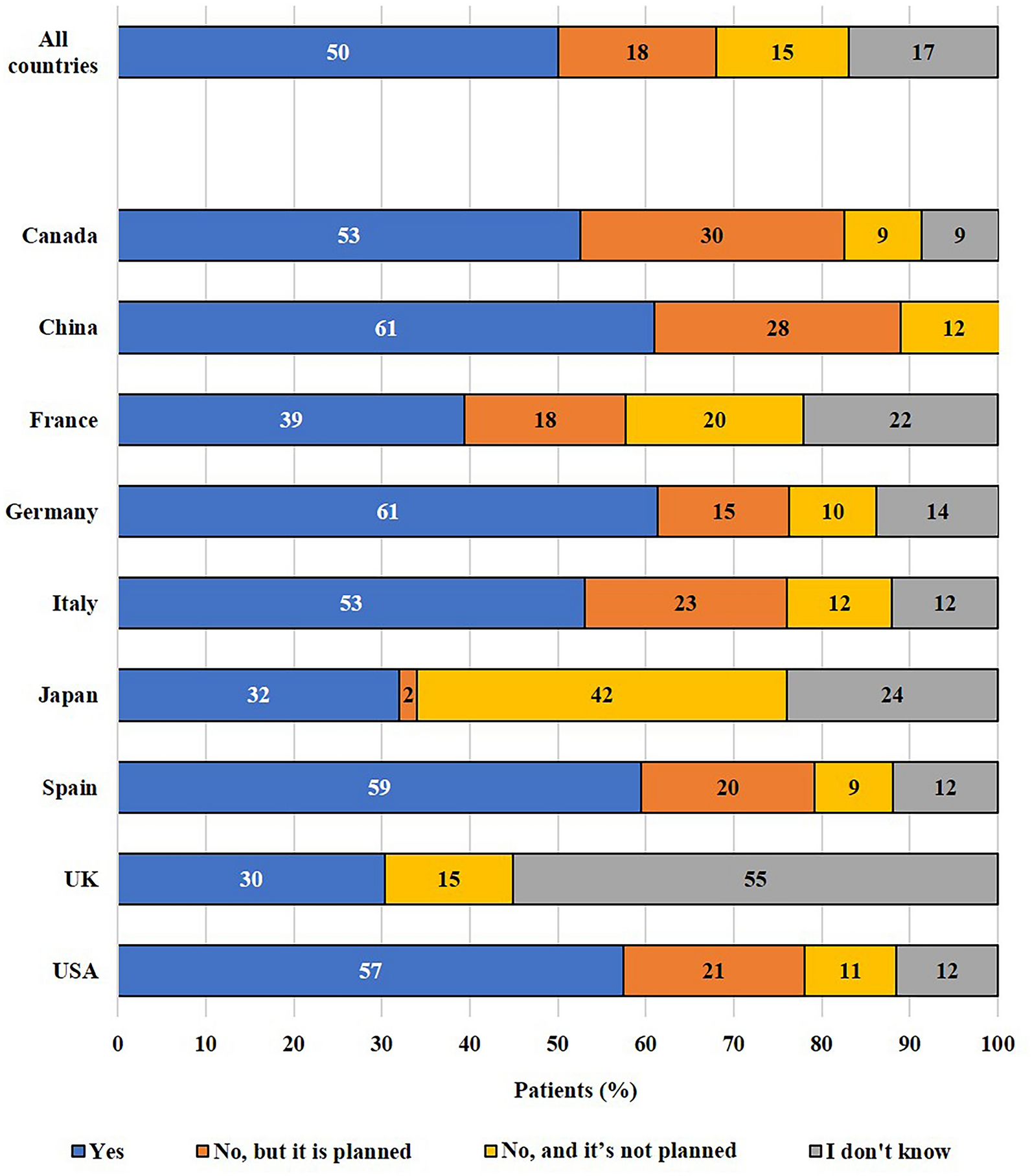

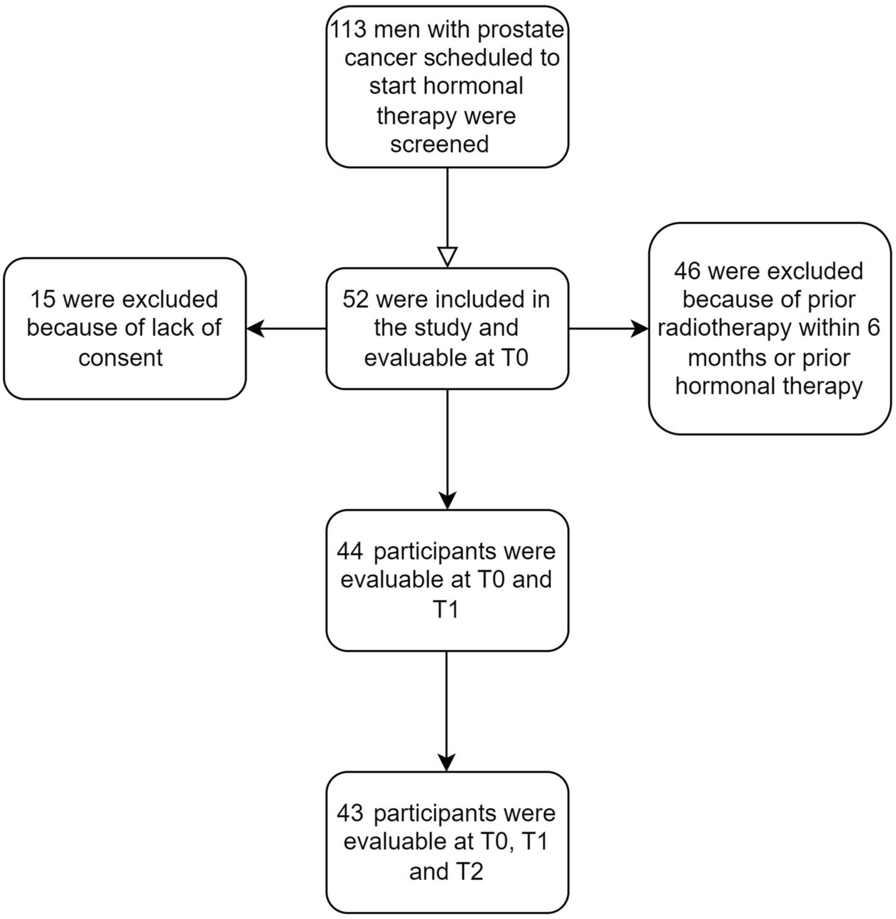

This was a phase 1, randomized, double-blind, three-way parallel-group clinical trial conducted in two centers in New Zealand between July 2021 and May 2023. The drugs used in this trial were BP02 (CuraTeQ Biologics Private Limited, India), reference EU-trastuzumab (Herceptin®, Roche Pharma AG, Grenzach-Wyhlen, Germany), and reference US-trastuzumab (Herceptin®, Genentech, Inc., South San Francisco, CA, USA).
The trial was restricted to healthy male volunteers aged 18 to 65 years to control variability. Subjects were eligible for study inclusion if they had a BMI (body mass index) of 18–30 kg/m2, a body weight of 50–100 kg, and had no clinically relevant abnormalities of any body system based on history, physical examination, and laboratory tests, including thyroid function tests, 12-lead electrocardiogram (ECG), and echocardiogram. Subjects were required to refrain from donating sperm or fathering a child or they agreed to use two acceptable contraceptives for up to 9 months after the administration of the test drug. Subjects were also required to abstain from smoking for 48 h prior to recruitment.
Subjects with a known history of hypersensitivity to trastuzumab, those who had any clinically relevant condition of any organ system detected by a history check, physical examination, ECG, or laboratory investigation, or those who had a left ventricular ejection fraction (LVEF) < 55% were excluded from the study. Other major exclusion criteria were a history of cancer; testing positive for HIV, HBsAg, or HCV; the concurrent use of any drug or supplement that, in the opinion of the investigator, had the potential to interfere with the study results; the receipt of a COVID-19 vaccine within 14 days prior to or within 15 days after the completion of the study; the use of hematopoietic growth factors, monoclonal antibodies, or immunoglobulins within 6 months prior to screening (or five half-lives, whichever was longer); major surgery or trauma within the past 1 year; a history of or current alcohol abuse or drug abuse; and the receipt of any monoclonal antibody therapy as a part of a study within 90 days prior to dosing. The complete list of inclusion and exclusion criteria is available in Supplementary Table 1 in the Supplementary Information.
Eligible subjects were randomized 1:1:1 to receive a single 6-mg/kg dose of BP02, EU-trastuzumab, or US-trastuzumab via IV infusion over 90 min. Dosage selection was based on the approved maintenance dose of 6 mg/kg over 90 min for various indications [11] and similar doses used in previous studies [12, 13]. Randomization was carried out using a number list within coded envelopes and in a double-blind fashion, with treatment allocation concealed until the database was locked after study completion.
Outcomes and AssessmentsPharmacokinetic EvaluationsBlood samples were collected immediately pre-infusion (0 min); post-dose samples were taken 0.5, 1, 4, and 6 h after the end of infusion (±10 min window), at 24 h and 48 h post-dose relative to the start of infusion, and on days 3, 4, 8, 15, 22, 29, 36, 50, 64, and 78 (Fig. 1). A validated indirect sandwich enzyme-linked immunosorbent assay (ELISA) method was used to measure trastuzumab in human serum [14].
Fig. 1
Study design. Healthy men were randomized to receive a single 90-min infusion of 6 mg/kg of BP02, US-trastuzumab, or EU-trastuzumab; blood samples were collected at pre-specified intervals. aOne subject who was randomized to EU-trastuzumab withdrew their consent prior to dosing. bReasons for discontinuation post-discharge: BP02, AEs (N = 1); US-trastuzumab, AEs (N = 3) and withdrawal of consent (N = 1). AEs adverse events
The primary PK endpoints were area under the serum concentration–time curve (AUC) from the time of dosing to infinity (AUC0-inf), AUC from the time of dosing until the time of the last quantifiable concentration (AUC0-t), and peak serum concentration of trastuzumab (Cmax). Secondary PK endpoints were time to achieve Cmax (Tmax), half-life (T½), elimination rate constant (λz), and systemic clearance of trastuzumab (CL). The PK similarity of EU-trastuzumab and US-trastuzumab was also evaluated to provide bridging data that can justify the use of a single reference product in future comparative clinical trials in patient populations.
Safety EvaluationsSafety evaluations were performed based on physical examination, vital signs, ECG parameters, echocardiogram, clinical safety laboratory tests (hematology, clinical biochemistry, coagulation, urinalysis, and urine microscopy where clinically indicated), infusion-related reactions, adverse events (AEs), and concomitant medication.
All safety evaluations were conducted at screening, at subject discharge from the unit, and at study exit or early termination. Vitals were checked and a physical examination was conducted at every subject contact; a 12-lead ECG and an echocardiogram were conducted on days 8, 22, and 78, with two additional ECG recordings made on days 3 and 4; blood and urine analyses were performed on days 2, 4, 8, 15, 29, 50, and 78.
AEs were coded using a central coding dictionary, the Medical Dictionary for Regulatory Activities (MedDRA), version 25.1. An AE was defined as any clinically significant observation outside the normal range, and a treatment-emergent AE (TEAE) was defined as any AE that first occurred or worsened in severity after the administration of the study treatment.
Immunogenicity EvaluationsBlood samples were collected for the measurement of antidrug antibodies (ADA) and neutralizing antibodies (NAb) on days 1, 50, and 78. A validated electrochemiluminescence immunoassay (ECLIA) method was used for the determination of trastuzumab anti-drug antibodies and neutralizing anti-drug antibodies in human serum [15].
Subjects who were confirmed positive for anti-trastuzumab antibodies, including subjects who discontinued the study after receiving the drug, were followed up for up to 12 months. These subjects attended the clinical center at 6, 9, and 12 months post-dose for blood sampling to confirm their immunoglobulin status or until two consecutive samples had been confirmed negative for anti-trastuzumab antibodies, whichever occurred earlier.
Statistical AnalysisPK parameters were derived using non-compartmental methods with Phoenix® WinNonlin® version 8.3 (Certata, L.P., Princeton, NJ, USA). Cmax was obtained directly from the observed concentration versus time data, and Tmax was obtained from Cmax. AUC0-t was calculated by linear-up/log-down trapezoidal summation; AUC0-inf was calculated as the sum of AUC0-t plus the ratio of the last measurable plasma concentration to the elimination rate constant; and CL was calculated as dose divided by AUC0-inf. λz was calculated from a semi-log plot of the serum concentration–time curve by linear least-squares regression analysis using the maximum number of points in the terminal log-linear phase, and T½ was calculated as 0.693/λz.
The general linear models procedure (PROC GLM) of the SAS software (SAS Institute, Cary, NC, USA) was used for statistical analyses. If the 90% confidence intervals (CIs) for AUC0-inf, AUC0-t, and Cmax were within the 80–125% range (natural-log-transformed data, one-sided tests, α = 0.05), the comparison was considered to indicate bioequivalence.
Considering an expected treatment ratio of 95%, a maximum observed inter-subject coefficient of variation of approximately 20%, and a dropout rate of approximately 10%, a sample size of 37 subjects per arm was calculated as providing at least 90% power at a 5% significance level for all pairwise comparisons for the primary endpoints.
Ethical Conduct in the StudyThis study was conducted in accordance with the ethical principles enshrined in the Declaration of Helsinki as well as all applicable local and regional laws. All subjects provided informed consent prior to study initiation. The study was approved by the Health and Disability Ethics Committee (HDEC; approval number 20/STH/190) and was registered at the Australian New Zealand Clinical Trials Registry (ANZCTR) prior to the initiation of subject screening (ACTRN12621000573853).
留言 (0)