
記住我


To explore the role of VC on TSCs differentiation, the primary mouse TSCs underwent culture in a differentiation medium with varying VC concentrations (10, 30, or 100 μM) for 3 days. Cell viability assessments using the MTT assay unveiled a dose-dependently enhancement in cell viability with VC treatment (Fig. S1A). Morphological assessments through light microscopy revealed that TSCs cultured in differentiation medium displayed larger cell sizes and blurred boundaries compared to those cultured in proliferation medium, indicating a transition toward TGC differentiation. Notably, the presence of VC further amplified these morphological alterations, yielding cells with enhanced irregularities and a network-like appearance, indicative of an advanced stage of TGC differentiation (Fig. S1B).
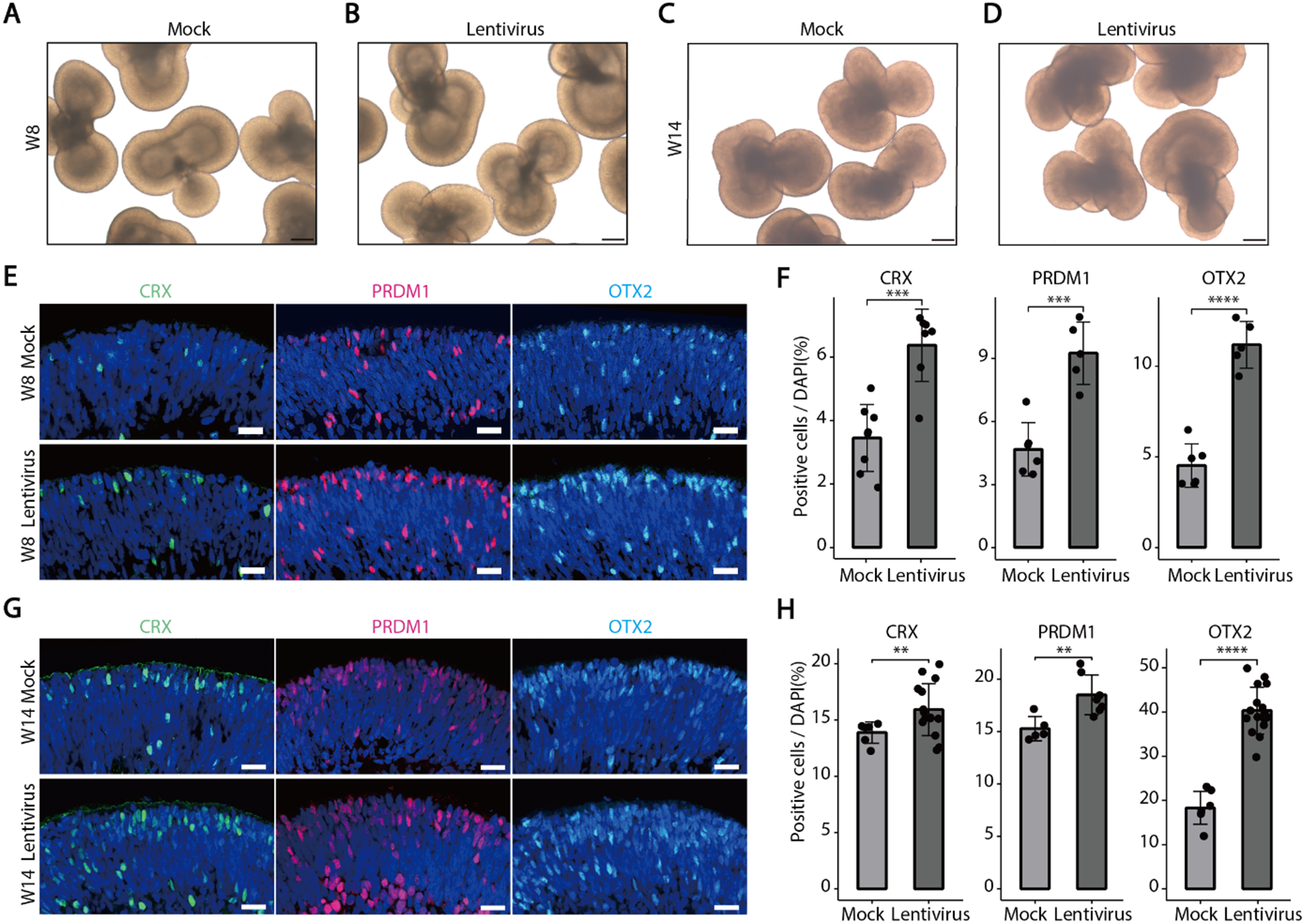
Then, quantitative RT-PCR (qPCR) analysis of trophoblast-specific markers, including P-TGC markers (P450scc, Plf, PL-1, PL-2), S-TGC marker Ctsq, and SpTs marker Tpbpa, as well as ST-I marker (SynA) and ST-II marker (SynB), indicated that VC treatment robustly induced the mRNA levels of Ctsq, PL-2, PL-1, P450scc, and Plf by approximately 4.7-, 2.7-, 1.2-, 0.8- and 0.4-fold, respectively, compared to vehicle treatment (Fig. 1A–C). Additionally, VC treatment enhanced Tpbpα mRNA levels by 1.3-fold, with no discernible impact on SynA or SynB mRNA levels (Fig. 1B, C). All these findings collectively highlight VC as a potent driver of TSCs differentiation, particularly into diverse TGC subtypes.
Fig. 1
VC induces the TSC differentiation and Hand1 expression. A-C Quantitative RT-PCR assays for trophoblastic differentiation markers in the primary TSCs incubated in the differentiation medium with or without VC at 100 μM for 3 days. P-TGC markers, P450scc, Plf, PL-1, PL-2; S-TGC marker Ctsq; SpTs marker Tpbpa; ST-Is and ST-IIs markers SynA and SynB, respectively. D-E Western analyses of Hand1 in whole primary TSCs or in cytosolic versus nuclear fractions of primary TSCs incubated in the differentiation medium with or without VC for 48 h. F–H Western and immunofluorescence analyses of the Flag tag in JEG-3 cells transfected with Flag-Hand1 and treated with VC for the indicated times or 48 h. Numerical data: mean ± SD, n = 3, *p < 0.05, **p < 0.01
Given the pronounced enhancement of TGC markers in response to VC treatments, and Hand1's integral role in the differentiation of all TGC subtypes [5], the Hand1 mRNA and protein levels in TSCs following VC exposure were evaluated using qPCR and Western blot analysis. We found that VC treatments did not alter the Hand1 mRNA levels but increased the Hand1 protein expression in a dose-dependent manner, particularly treated with 30 and 100 μM VC (Fig. 1D and Fig. S1C). Subsequent Western blot analysis of cytosolic and nuclear fractions of Hand1 protein corroborated heightened cytosolic and nuclear Hand1 levels post-VC treatments (Fig. 1E). Likewise, further assessments conducted in human trophoblast-like choriocarcinoma JEG-3 cells or HEK293T cells (293T) transiently transfected with Flag-tagged Hand1 (Flag-Hand1) consistently elicited a dose- and time-dependent increase in exogenous Hand1 protein levels upon VC treatments (Fig. 1F, G) and Fig. S1D). And immunofluorescence analysis of Flag-Hand1 in JEG-3 cells further demonstrated increased exogenous Hand1 levels in both the cytoplasm and nucleus in the presence of VC (Fig. 1H). The above results underscore the pivotal role of VC in enhancing Hand1 protein expression during TSCs differentiation.
Basic HLH factors typically form homodimers or heterodimers to induce TGC differentiation, and induction of TSCs differentiation into TGCs requires the formation of Hand1 homodimers for transcription regulation [24]. To determine if VC modulates the formation of Hand1 homodimers, a co-immunoprecipitation assay was performed in JEG-3 cells co-transfected with HA-Hand1 and Flag-Hand1 and treated with or without VC. Western assays using a Flag antibody indicated that VC treatment did not affect the Hand1 homodimerization but rather augmented the quantity of Hand1 homodimers by eliciting an increase in Hand1 protein expression (Fig. S1E). These findings suggest VC induces TSCs differentiation into TGC subtypes primarily by upregulating Hand1 expression rather than modulation of its dimerization capacity.
VC suppresses JNK signaling to enhance Hand1 expression for TSCs differentiation into TGCsSince VC is recognized for its potent antioxidant properties in cellular functions, we investigated whether VC induces TSCs differentiation into TGCs by leveraging its antioxidative capabilities. TSCs were respectively incubated with various antioxidants such as N-acetyl cysteine (NAC), α-tocopherol (VE), glutathione (GSH), and lipoic acid (LA), and the impacts of these antioxidants on Ctsq mRNA expression were assessed using qPCR analyses, juxtaposing their induction effects with that of VC. Intriguingly, while VC treatment notably elevated Ctsq mRNA levels, other antioxidants like NAC, VE, GSH, and LA exhibited no noteworthy impact on basal Ctsq mRNA expression. Conversely, simultaneous treatment of VC with different oxidants such as H2O2 or Diethyl maleate (DEM) failed to counteract VC’s enhancement of Ctsq mRNA expression (Fig. S2). Based on these results, we speculated that VC induces TSCs differentiation into TGCs independent of its anti-oxidative property.
In a previous study, it was demonstrated that fibroblast growth factor 4 (FGF4)-mediated maintenance of TSCs stemness relies on MAPK kinase kinase (MEKK4)-triggered JNK signaling [25], whereas our previous research revealed that DHA, an oxidized form of VC, could inhibit JNK phosphorylation to regulate steroidogenesis in human trophoblast-like JAR cells [26], hinting at potential effects of VC on JNK signaling. To identify the impact of VC on TSCs’ MAPK signaling, Western analyses revealed that exposure of TSCs to VC for 30 min led to a significant reduction in phosphorylated JNK (p-JNK) and P38 (p-P38) levels by up to 30% and 40%, respectively, while leaving phosphorylated extracellular signal–regulated kinases 1/2 (p-ERK) unaffected (Fig. 2A). In contrast, anisomycin, a dual JNK and P38 agonist, time-dependently negated Hand1 protein levels and dose-dependently diminished VC-induced Ctsq mRNA in TSCs (Fig. 2B, C). Noteworthy, the JNK inhibitor SP600125 dose-dependently increased Ctsq mRNA levels in TSCs, whereas the P38 inhibitor SB203580 had no discernible effect on Ctsq mRNA levels (Fig. 2D, E). Furthermore, anisomycin and SP600125 markedly lowered and raised Flag-Hand1 expression in JEG-3 cells, respectively, whereas SB203580 had no impact on Flag-Hand1 levels (Fig. 2F, G). These results suggest that JNK signaling, rather than P38 and ERK1/2 signaling, plays a pivotal role in VC-induced Hand1 protein expression and the differentiation of TSCs into TGCs.
Fig. 2
VC inactivates JNK to stabilize Hand1. A Western blot analysis of MAPK activity in the primary TSCs incubated with or without VC for 30 min. B Western analyses of Hand1 in the primary TSCs incubated with or without anisomycin at 10 μM. C-E Quantitative RT-PCR assays of Ctsq in the primary TSCs incubated with or without anisomycin, SB203580 or SP600125 and in the presence or absence of VC for 72 h. F-G Western analyses of Flag tag for JEG-3 cells transfected with Flag-Hand1 and treated with anisomycin for 24 h or 10 μM SB203580(SB20)/25 μM SP600125(SP60) for 12 or 24 h. H-I Western analyses of the Flag tag in JEG-3 cells transfected with Flag-Hand1 and JNK1* and treated with or without VC for 24 h. J Western analyses of the Flag tag in JEG-3 cells transfected with Flag-Hand1 for 48 h, following the infection with Scramble- or JNK1/2-shRNA-expressing lentiviruses. K Western analyses of Flag-p-JNK1 for the immunocomplex precipitated by a Myc or Flag antibody in JEG-3 cells transfected with Flag-JNK1* and Myc-Hand1. L Western analyses of Flag tag (15% SDS-PAGE) in JEG-3 cells transfected with Flag-JNK1*/2* and Flag-Hand1 and treated with MG132 at 10 μM or DMSO for 24 h. M Western analyses of HA tag for the immunocomplex precipitated by a Flag antibody in JEG-3 cells transfected with Flag-Hand1 and HA-ubiquitin and treated with or without 100 μM VC in the presence of 10 μM MG132. Numerical data: mean ± SD, n = 3, *,+p < 0.05, **,++p < 0.01
To further elucidate the involvement of JNK signaling in VC-induced Hand1 expression, gain- and loss-of-function experiments of JNK signaling were executed by transfecting JEG-3 cells with a constitutively active form of JNK1 (JNK1*) plasmid created through a fusion protein of Jun N-terminal kinase kinase 2 (JNKK2) and JNK1, as well as lentiviral transduction to knock down JNK1/2. We found that JNK1* overexpression significantly diminished both basal and VC-induced Flag-Hand1 protein expression levels in JEG-3 cells, while JNK1/2-shRNA-expressing lentiviruses, which reduced the JNK1/2 by approximately 80%, increased Flag-Hand1 protein levels by 2.3-fold, compared with Scramble-shRNA-expressing lentiviruses (Fig. 2H–J). Then, to probe the potential interaction between Hand1 and JNK1*, co-immunoprecipitation experiments were carried out using cell lysates from JEG-3 cells transiently transfected JNK1* alone, Myc-Tag Hand1 (Myc-Hand1) alone, or both plasmids together. We found that cells expressing either Myc-Hand1 or JNK1* in isolation showed limited JNK1*, whereas co-expression of both proteins exhibited abundant JNK1* (Fig. 2K). Therefore, JNK1* physically interacts with and negates the Hand1 protein.
To ascertain whether JNKs negate the Hand1 protein through proteasomal degradation pathway, JEG-3 cells expressing Flag-Hand1 and JNK1*/JNK2* were treated with MG132, a proteasome inhibitor, and Western analyses on a 15% SDS-PAGE were utilized to assess potential shifts in Hand1 band post-JNK-mediated phosphorylation. The results revealed that both JNK1* and JNK2* robustly reduced Flag-Hand1 levels in JEG-3 cells, while JNK1* exhibited stronger effects than JNK2* (Fig. 2L). Moreover, JNK1* and JNK2* consistently caused an apparent upward shift in Flag-Hand1 bands, while MG132 treatments did not alter the band shift but effectively reversed the Hand1 reduction induced by JNK1* or JNK2* (Fig. 2L), suggesting a direct involvement of JNK1/2 in the degradation of Hand1 through the proteasomal manner. To confirm this notion, we used MG132 and VC to block the proteasomal degradation and JNK activity, respectively, in JEG-3 cells transfected with Flag-Hand1 and HA-tagged ubiquitin (HA-Ubiquitin). Co-immunoprecipitation utilizing a Flag antibody followed by Western blotting with a HA antibody was performed and the results indicated that VC-treated cells exhibited a 50% reduction in ubiquitinated Hand1 with a molecular weight prediction of 37 kDa (band #1) and a 30% decrease in ubiquitinated Hand1 with a molecular weight prediction of 50 kDa (band #2) compared to control cells (Fig. 2M). These findings suggest that VC inactivates JNK to block the ubiquitination-mediated proteasomal degradation of Hand1.
JNK phosphorylates Ser48 of Hand1 to facilitate its degradationThe interaction between JNK and Hand1 prompted a direct exploration of JNK's Hand1 phosphorylation. Utilizing the Group-based Prediction System (GPS), potential JNK phosphorylation sites on Hand1 were identified. Analysis of the Hand1 protein sequence unveiled high or low prediction scores for Ser33, Ser48, and Ser81 sites, as well as for Ser98, Thr107, and Ser109 sites, all conserved among human, mouse, and rat (Fig. 3A). To further investigate significance of these sites, Flag-Hand1 variants were generated with mutations at the consensus serine or threonine residue (Ser and Thr to Ala) individually (S33A, S48A, S81A, S98A, T107A, and S109A) and evaluated for their response to JNK1* in JEG-3 cells through Western analyses on a 10% SDS-PAGE. In the absence of JNK1*, Flag-Hand1 levels for the wild-type (WT) resembled those of S33A, S81A, S98A, T107A or S109A variants, but were 50% lower than the S48A variant; in the presence of JNK1*, Flag-Hand1 levels in WT persisted similar to S33A, S81A, S98A, T107A, or S109A variants, but were 85% lower compared to the S48A variant (Fig. S3A and B). Thus, Ser48 emerged as a key amino acid residue in mediating JNK-induced destabilization of Hand1.
Fig. 3
JNK directly phosphorylates Hand1 at Ser48 to destabilize Hand1. A The conserved sites of Hand1 for potential phosphorylation by MAPK. B Western analyses of Flag tag (15% SDS-PAGE) in JEG-3 cells transfected with Flag-Hand1 variants and Flag-JNK1*. Ratios: phosphorylated versus non-phosphorylated Flag-Hand1. C In vitro phosphorylation of GST-Hand1 variants by JNK2α2. Autoradiography signals were normalized to levels of GST-Hand1 variants. D-E Western analyses of Flag tag in JEG-3 cells transfected with Flag-Hand1 variants with or without the control EGFP-expressing vector in the presence or absence of 100 μM VC for 48 h. F Immunofluorescence assays of Flag tag in JEG-3 cells transfected with Flag-Hand1 variants and treated with or without VC for 48 h
Subsequently, Western assays were conducted on a 15% SDS-PAGE to assess phosphorylated and non-phosphorylated Flag-Hand1 bands following JNK1* overexpression. We found that the S33A and S48A variants displayed comparable levels of phosphorylated and non-phosphorylated Flag-Hand1 levels similar to the WT in the absence of JNK1*. Conversely, the S81A variant showed an unexpected elimination of phosphorylated Flag-Hand1 band while maintaining consistent levels of non-phosphorylated Flag-Hand1 band (Fig. 3B). In the presence of JNK1*, the WT, S33A, and S81A variants exhibited nearly identical ratios between phosphorylated and non-phosphorylated Flag-Hand1 levels (3:1), whereas the S48A variant markedly shifted this ratio to 1:5. Notably, both the S48A and S81A variants seemingly eliminated the highest phosphorylated band of Flag-Hand1 (Fig. 3B). These observations imply that while Ser81 on Hand1 protein may potentially serve as a phosphorylation site for kinases other than JNK, Ser48 remains crucial as a JNK-specific phosphorylation site.
To ascertain whether JNK could directly phosphorylate Hand1 at Ser48, an in vitro phosphorylation assay was conducted employing a commercially available active JNK2α2 with purified recombinant glutathione S-transferase (GST)-tagged WT Hand1 or its S48A mutant (Fig. S3C). Following SDS-PAGE and autoradiography, the WT Hand1 exhibited notable 32P incorporation, while the S48A variant showed no 32P uptake even in the presence of active JNK2α2 (Fig. 3C, top). Subsequent Western blotting with the GST antibody confirmed the 32P-labeled band corresponded to the GST-Hand1 variants, displaying consistent levels across reactive mixtures (Fig. 3C, bottom), indicative of direct phosphorylation of Hand1 by JNK at Ser48.
To evaluate the significance of Ser48 phosphorylation of the Hand1 protein, we introduced mutations by substituting the serine residue at position 48 of Hand1 with either the phosphomimetic aspartate (D) to stimulate activation of Hand1 or with alanine to mimic inactivation of Hand1, which was transfected into JEG-3 cells alongside an enhanced green fluorescent protein (EGFP) vector to monitor transfection efficiency. Western analyses confirmed consistent EGFP expression levels, ensuring uniform transfection effectiveness across samples. Notably, cells transfected with S48A and S48D mutants respectively exhibited a 150% increase and a 50% decrease in Flag-Hand1 levels compared to those transfected with the WT Hand1 (Fig. 3D). Moreover, treatment with 100 μM VC elicited a 1.4-fold elevation in Flag-Hand1 expression in cells transfected with WT construct, while no significant alteration was observed in cells transfected with the S48A or S48D variants (Fig. 3E). In parallel, immunofluorescent analysis of JEG-3 transfected with these Flag-Hand1 variants using Flag antibody revealed that, under basal conditions, cells transfected with WT Hand1 exhibited a relatively modest fraction of Hand1 in cytoplasm and an exclusive nucleus level; the Hand1 S48A mutation resulted in a much higher nucleus fraction compared to WT, whereas the Hand1 S48D mutation led to notably reduced levels of Hand1 in both the nucleus and cytoplasm. Moreover, VC stimulation promoted increased levels of WT Hand1 in both compartments, but not in those transfected with S48A or S48D variants (Fig. 3F). These in vitro findings underscore the crucial role of Ser48 in Hand1 for the VC-mediated enhancement of Hand1 protein expression.
The VC/JNK/Hand1 pathway induces TGC differentiation in murine placentasSince VC could suppress JNK activity to stabilize Hand1 expression, and the differentiation of TSCs into all TGC subtypes relies on the contribution of Hand1 protein [24, 27, 28], we further elucidated the mechanisms of VC/JNK/Hand1 pathway on TGC differentiation in E8.5 placentas by leveraging multiple strategies, including VC deficiency, lentivirus-mediated knockdown of JNK or overexpression of Hand1 mutants. We first generated global L-gulono-γ-lactone oxidase (Gulo−/−) knockout mice, genetically incapable of synthesizing VC, and raised on tap water containing 4 g/L of VC for normal growth [29]. We then crossed the Gulo−/− females with the Gulo−/− males to generate the Gulo-/- placentas, and pregnant Gulo−/− females were deprived of VC post-plug identification (E0.5). HPLC analysis of maternal serum VC concentrations indicated VC deprivation resulted in a progressive decline in serum VC concentrations, dropping from 53.5 μM from E0.5 to approximately 11.5 μM by E8.5 (Fig. S4A).
To investigate the VC deficiency on early development of placentation, pregnant Gulo−/− females were dissected at E8.5, and conceptuses were then subjected to cryosection. And HE staining and in situ hybridization with Pl-1 probe, a specific marker for P-TGC, which also serves as an indicator of Hand1 activity [6], were carried out. The results showed that in the presence of VC supplementation, Pl-1-positive P-TGCs distinctly lined implantation sites, demarcating the maternal decidua from the ectoplacental cone. Conversely, in the absence of VC supplementation, Pl-1-positive P-TGCs were faintly arrayed along implantation sites, fuzzily separating the maternal decidua from the ectoplacental cone (Fig. 4A, B), indicating VC insufficiency impairs P-TGCs differentiation at the implantation sites, potentially by inhibiting Hand1 activity.
Fig. 4
Pl-1 expression in the implantation sites of E8.5 placentas. A-B HE staining (A) and in situ hybridization assays of Pl-1 (B) in the implantation sites of E8.5 Gulo−/− placentas supplemented with or without VC. C-D HE (C) and in situ hybridization assays of Pl-1 (D) in the implantation sites of E8.5 placentas infected with Scramble- or JNK1/2-shRNA expressing lentiviruses. E GFP expression in the E3.5 blastocysts 8 h after infection with GFP-bearing and Hand1 variants-expressing lentiviruses. F-G HE staining (F) and in situ hybridization of Pl-1 (G) in the implantation sites of E8.5 placentas infected with GFP-bearing and Hand1 variants-expressing lentiviruses
To confirm the importance of JNK activity in TGC differentiation in vivo, E3.5 blastocysts were transduced with lentiviruses carrying green fluorescent protein (GFP) along with Jnk1/2-shRNA to specific knockdown JNK1/2 protein in trophectoderm, followed by blastocyst transplantation. Immunofluorescent analysis of GFP expression at E3.5 blastocysts indicated the lentiviruses were efficiently delivered to the trophectoderm, excluding the inner cell mass (ICM) (Fig. S4B). Moreover, Western blot analysis of the whole E16.5 placentas showed that Jnk1/2-shRNA, which reduced the JNK2 and JNK1 expression by approximately 36% and 63%, respectively, resulted in approximately a 2.5-fold elevation in the Hand1 protein levels compared to Scramble-shRNA (Fig. S4C). Consistently, in situ hybridization of Pl-1 indicated that Jnk1/2-shRNA robustly amplified the number of Pl-1-positive P-TGCs at E8.5 placentas, distinctly lining the implantation sites and clearly separating the maternal decidua from the ectoplacental cone, in contrast to Scramble-shRNA (Fig. 4C, D). These results collectively demonstrate that JNK inactivation enhances TGC differentiation in murine placentas by increasing both Hand1 protein levels and activity.
Then, the in vivo significance of Hand1 mutants on TGC differentiation was assessed by transducting E3.5 blastocysts with GFP-carrying lentiviruses expressing Hand1(WT)-, Hand1(S48A)- or Hand1(S48D), respectively, followed by transplantation. Immunofluorescent analysis of GFP expression of E3.5 blastocysts prior to transplantation revealed that the GFP-carrying Hand1 variants were effectively integrated and uniformly expressed in the trophectoderm (Fig. 4E). Moreover, HE staining and in situ Pl-1 hybridization revealed that overexpression of Hand1(WT) moderately enhanced the number of Pl-1-positive P-TGCs; overexpression of Hand1(S48A) robustly enhanced the number of Pl-1-positive P-TGCs at E8.5 implantation sites, whereas overexpression of Hand1(S48D) notably reduced the number of Pl-1-positive P-TGCs compared to Hand1(WT) overexpression (Fig. 4F, G), suggesting S48A and S48D mutations in Hand1 have positive and negative effects on TGC differentiation. Overall, these in vivo findings highlight that VC inactivates JNK signaling to increase the Hand1 protein level and activity, thereby facilitating TGC differentiation in placentas.
VC deficiency causes the failure of maintenance of pregnancySubsequently, we dissected the significance of VC in sustaining pregnancy by studying the midgestational (E12.5 and E14.5) to late-gestational stages (E16.5 and E18.5) of pregnant Gulo-/- females carrying Gulo-/- progeny. We found VC withdrawal since plug identification (E0.5) led to a substantial reduction of maternal serum VC levels by approximately 94% persisting through E12.5~E18.5, in comparison to 4 g/L VC supplementation (Fig. S4D). Then, the effects of VC deficiency on fetal and placental development were first assessed by determining the placental and fetal numbers and the corresponding results unveiled that VC deprivation in the pregnant Gulo-/- females did not significantly alter the numbers of implanted embryos and placentas, however, VC deficiency markedly escalated the incidence of atrophic and absorbed placentas and embryos by approximately 7%, 19%, and 38% at E14.5, E16.5, and E18.5, respectively, exhibiting no significant effect at E12.5, as compared to VC supplementation (Fig. 5A, B). Furthermore, VC deficiency consistently reduced the weights of both viable placentas (data not shown) and embryos by around 10%, 22%, and 30% at E14.5, E16.5, and E18.5, respectively (Fig. S5B-D). Consequently, the inadequacy of VC fails to sustain pregnancy in Gulo-/- matrices bearing Gulo-/- offspring.
Fig. 5
VC deficiency fails to maintain the pregnancy. A-B Numbers of the implanted and absorbed placentas and embryos from the pregnant Gulo−/− females (crossed with Gulo−/− males) supplemented with or deprived of VC at 4 g/L in the tap water from E0.5. Specifically, we analyzed a total of 245 placentas, distributed as follows: 61 placentas each at E12.5, E14.5, and E16.5, and 62 placentas at E18.5. In the VC-deficient group, the incidence of affected placentas was approximately 3% (n = 1) at E12.5, 7% (n = 2) at E14.5, 19% (n = 6) at E16.5, and 38% (n = 12) at E18.5. In contrast, the VC-supplemented group showed significantly lower incidences: 3% (n = 1) at each of E12.5, E14.5, and E16.5, and 7% (n = 2) at E18.5. Arrows: absorbed placentas and embryos. C-E Western and immunohistochemistry assays of Hand1 or p-JNK in the viable placentas with the same treatments as described in (A and B). F-G VC concentrations in the sera and embryos of pregnant Gulo−/− females (crossed with WT males) supplemented with or deprived of VC at 4 g/L in the tap water from E0.5. H Numbers of the implanted and absorbed placentas from the pregnant Gulo−/− females as described in (F and G). Numerical data: mean ± SD, n = 6 ~ 8, *p < 0.05, **p < 0.01
Osteogenic disorder (ODS) rats (genotype od/od) manifest a deficiency in L-gulonolactone oxidase, leading to a hereditary defect in VC biosynthesis, necessitating a minimum of 1 g/L of VC supplementation for normal growth [30]. To reaffirm the role of VC in supporting pregnancy, we interbred the ODS rats and withheld VC upon plug identification. Consistent with findings from Gulo−/− mice, analysis of placentas and pups in ODS rats demonstrated that maternal VC deprivation notably increased the instances of atrophic and absorbed placentas and embryos by approximately 63% at E14.5 compared to VC supplementation, with no significant alterations in the quantities of implanted embryos and placentas being noted (Fig. S5A). As VC plays critical roles in the synthesis of the ovary and placental hormones that are essential for maintenance of pregnancy [13, 14], assessment of serum progesterone (P4) and estradiol (E2) levels in pregnant ODS rats at E14.5 revealed no significant difference between groups with VC supplementation or deprivation (Fig. S5B and C). Therefore, the inability of ODS rats with VC deficiency to sustain pregnancy is not attributed to insufficiency levels of P4 and E2.
Subsequent analysis concentrated on assessing the effects of VC deficiency on Hand1 and p-JNK1/2 levels in middle- and late-gestational stage of Gulo-/- placentas using Western blot analyses, and the outcomes indicated that VC deficiency reduced the Hand1 levels in placentas by ~50% at E12.5 and ~90% at E16.5 (Fig. 5C). In parallel, immunohistochemistry staining of Hand1 expression at E16.5 placental sections depicted that VC deficiency resulted in a notable decrease in Hand1 expression globally (Fig. 5D). In contrast, VC deficiency led to a notable increase in p-JNK1/2 levels by ~104% at E12.5 and ~175% at E16.5 Gulo-/- placentas (Fig. 5E). Thus, consistent with the aforementioned in vitro and in vivo findings, VC deficiency not only elevates p-JNK1/2 levels but also diminishes Hand1 levels in murine placentas.
To confirm the importance of fetal VC in maintaining pregnancy over maternal VC, Gulo-/- females were mated with WT males to generate Gulo+/- pups, with VC withheld upon plug identification. Interestingly, VC deprivation decreased the maternal serum VC levels to approximately 9 μM (versus ~60 μM) (Fig. 5F), and reduced VC levels of Gulo+/- offsprings to approximately 52 μM (versus ~59 μM) at both E16.5 and E18.5 compared to VC supplementation (Fig. 5G), indicating that in contrast to Gulo-/- pups, Gulo+/- pups resulting from the cross of female Gulo-/- and male Gulo+/+ mice can synthesize VC on their own despite a lack of VC supplementation to maternal Gulo-/- mice. In line with the VC levels observed in the Gulo+/- offspring, VC deprivation in Gulo-/- matrices bearing Gulo+/- pups did not result in a notable increase in the counts of the atrophic and adsorbed placentas and embryos, nor did it lead to a significant decline in the placental Hand1 expression, when compared with VC supplementation (Fig. 5Hand Fig. S5D). All these data collectively suggest that fetal VC deficiency, rather than maternal VC deficiency, is specifically linked to the failure of placentation and the maintenance of pregnancy.
VC deficiency disrupts trophoblastic differentiation in murine placentasFor a more in-depth understanding of the specific roles of VC in placentation, HE staining, immunohistochemistry staining, in situ hybridization, and transmission electron microscopic examination were performed using the viable Gulo−/− placentas at E16.5 and/or E18.5. HE staining results indicated consistent reductions in the dimensions of the placental mass, junctional zone, and labyrinthine layer at both E16.5 and E18.5, with minimal changes in the decidua basalis (Fig. 6A, B). While the LB layer of VC-supplemented Gulo−/− placentas exhibited a well-structured vascular network, that of VC-deficient Gulo−/− placentas displayed a disheveled and strip-shaped vascular network with dilated MBSs, expanded FBV spaces, and sparse trophoblastic layers separating the MBSs from FBV (Fig. 6A, B). Immunostaining of MBSs and FBV using antibodies against cytokeratin (CK) and laminin further demonstrated an augmented density of MBV and FBV in Gulo−/− placentas at E16.5 (Fig. 6C, D). The above results suggest that VC deficiency causes severe structural defects in the Gulo−/− placenta, potentially contributing to the failure of maintenance of pregnancy.
Fig. 6
VC deficiency disrupts the differentiation of TGCs during placentation. A-B HE staining of E18.5 and E16.5 placental sections from the pregnant Gulo−/− females (crossed with Gulo−/− males) and supplemented with ( +) or deprived of (-) VC at 4 g/L in the tap water from E0.5. C-D Immunohistochemistry of CK and Laminin in the placental labyrinths at E16.5. E Immunohistochemistry of P450scc and semi-quantification in the E14.5 placental sections. F-K In situ hybridization assays and semi-quantification in the E16.5 placental sections. L Transmission electron microscopic examination of trilaminar interhemal barrier of E16.5 placentas. Numerical data: mean ± SD, n = 4, **p < 0.01
Subsequently, in situ hybridization or immunohistochemistry staining was performed to detect the expression of trophoblastic markers in the Gulo−/− placentas. P450scc, a maker of P-TGCs, exhibited robust expression in E14.5 placentas supplemented with VC but sporadic expression in those lacking VC (Fig. 6E). VC deficiency led to a 59% reduction in Tpbpα-positive areas in the junctional zone of placentas compared to VC supplementation (Fig. 6F). Likewise, the areas positive for Plf-, Pl-II-, and Ctsq in the labyrinth of placentas decreased significantly by 47%, 48%, and 64%, respectively, compared to VC-supplemented Gulo−/− placentas (Fig. 6G–I), aligning with the in vitro findings (Fig. 1A–C) In addition, although the labyrinth expression of SynA, a marker of ST-I trophoblasts, seemingly remained unaffected by VC deficiency, the labyrinth expression of SynB, a marker of ST-II trophoblasts, notably increased with VC deficiency compared to VC supplementation (Fig. 6J, K), emphasizing compromised trophoblast functions. Finally, we examined the tri-laminar interhemal barrier (TIB) of the placental labyrinth via transmission electron microscopic. Labyrinths from VC-supplemented Gulo−/− placentas showed a typical TIB structure consisting of an S-TGC layer and two tightly adhered syncytiotrophoblast layers (ST-I and ST-II) lining the fetal endothelium and separating the MBL from FBV (Fig. 6I). Conversely, labyrinths from VC-deficient Gulo−/− placentas showed almost normal fetal endothelium with a contiguous basement membrane around ST-II, an atrophied S-TGC layer apparently lacking cell protrusions, an expanded ST-I layer adhering to S-TGC layer filled with dilated cytoplasmic vacuoles hindering intracellular transport, and an enlarged ST-II layer with excess lipid inclusions (Fig. 6I). Taken together, all these findings collectively suggest VC deficiency substantially activates JNK signaling to diminish Hand1 protein, facilitating developmental defects in trophoblasts within mouse placentas, particularly impacting the differentiation of multiple TGC subtypes, possibly accounting for the breakdown of maintenance of pregnancy.
留言 (0)